


Abstract
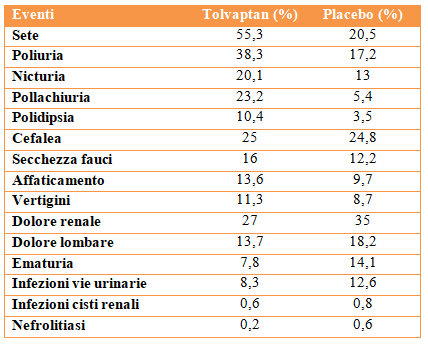
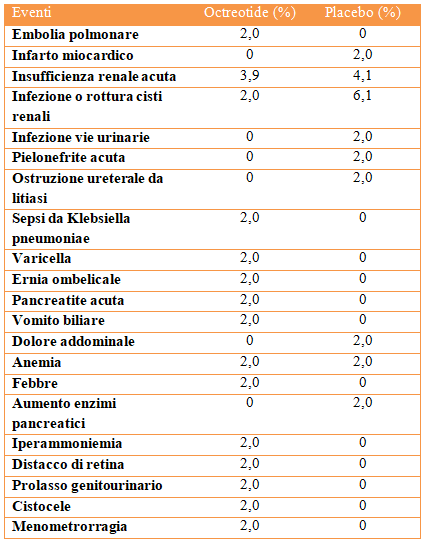
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is the most frequent monogenic hereditary disease as well as the most studied inherited kidney disease. Two drugs have recently been authorized that can slow down the progression of the disease: Tolvaptan (vasopressin receptor antagonist) and Octreotide-LAR (long-acting somatostatin analogue); they both are able to reduce the activity of cyclic adenosine monophosphate (cAMP) and therefore have anti-proliferative and anti-secretory effects. This review analyzes the main trials published to date demonstrating the effects on disease progression in patients with ADPKD and illustrates the indications for identifying subjects eligible for therapy.
Keywords: ADPKD, Tolvaptan, Octreotide
{kind=link}
{kind=link}
{kind=link}
{kind=link}