


Abstract
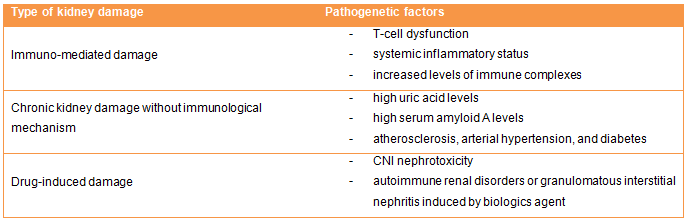
Kikuchi-Fujimoto disease (KFD), or Histiocytic Necrotizing Lymphadenitis, is a rare disease, with worldwide distribution but is best known in Japan and South Asia. The most common feature is cervical lymphadenopathy, accompanied by tenderness or high fever, with night sweats, but it can also be asymptomatic or with a very wide range of symptoms. The diagnosis is histopathological, on excisional biopsy. The Kikuchi-Fujmoto disease can mimic lymphoma but also tuberculosis and some autoimmune diseases, or be associated with them. Nephrologists need to be aware of it, considering the potential renal involvement. The association with systemic lupus erythematosus (SLE) is the most frequent but not the only one. Early diagnosis of this disease can prevent unnecessary investigations and aggressive therapies.
Keywords: Kikuchi-Fujimoto disease, Histiocytic Necrotizing Lymphadenitis, renal involvement, autoimmune disease, excisional biopsy, differential diagnosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}