


Abstract
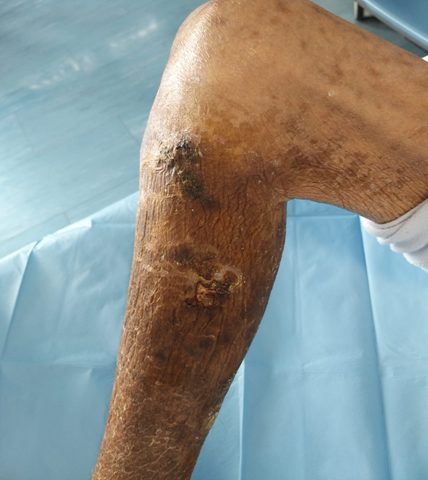
Primary hyperoxaluria (PH) is a rare genetic disorder with autosomal recessive transmission, characterized by high endogenous production and markedly excessive urinary excretion of oxalate (Ox). It causes the accumulation of calcium oxide crystals in organs and tissues including bones, heart, arteries, skin and kidneys, where it may cause oxalo-calcic nephrolithiasis, nephrocalcinosis and chronic renal failure. Some forms are secondary to enteric diseases, drugs or dietetic substances, while three primitive forms, caused by various enzymatic defects, are currently known: PH1, PH2 and PH3.
An early diagnosis, with the aid of biochemical and genetic investigations, helps prevent complications and establish a therapeutic strategy that often includes liver and liver-kidney transplantation, improving the prognosis of these patients.
In this work we describe the clinical case of a patient with PH1 undergoing extracorporeal hemodialysis treatment and we report the latest research results that could change the life of patients with PH.
Keywords: primitive hyperoxaluria, PH, nephrocalcinosis, chronic renal failure

{kind=link}
{kind=link}
{kind=link}
{kind=link}