


Abstract
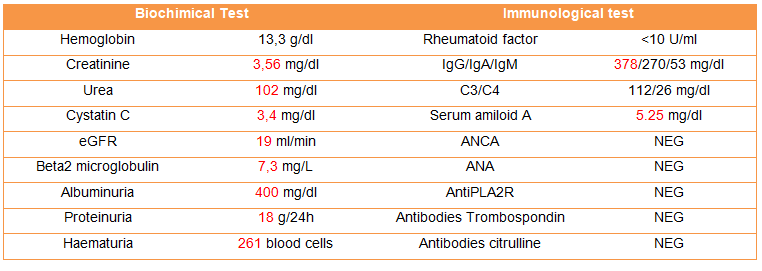
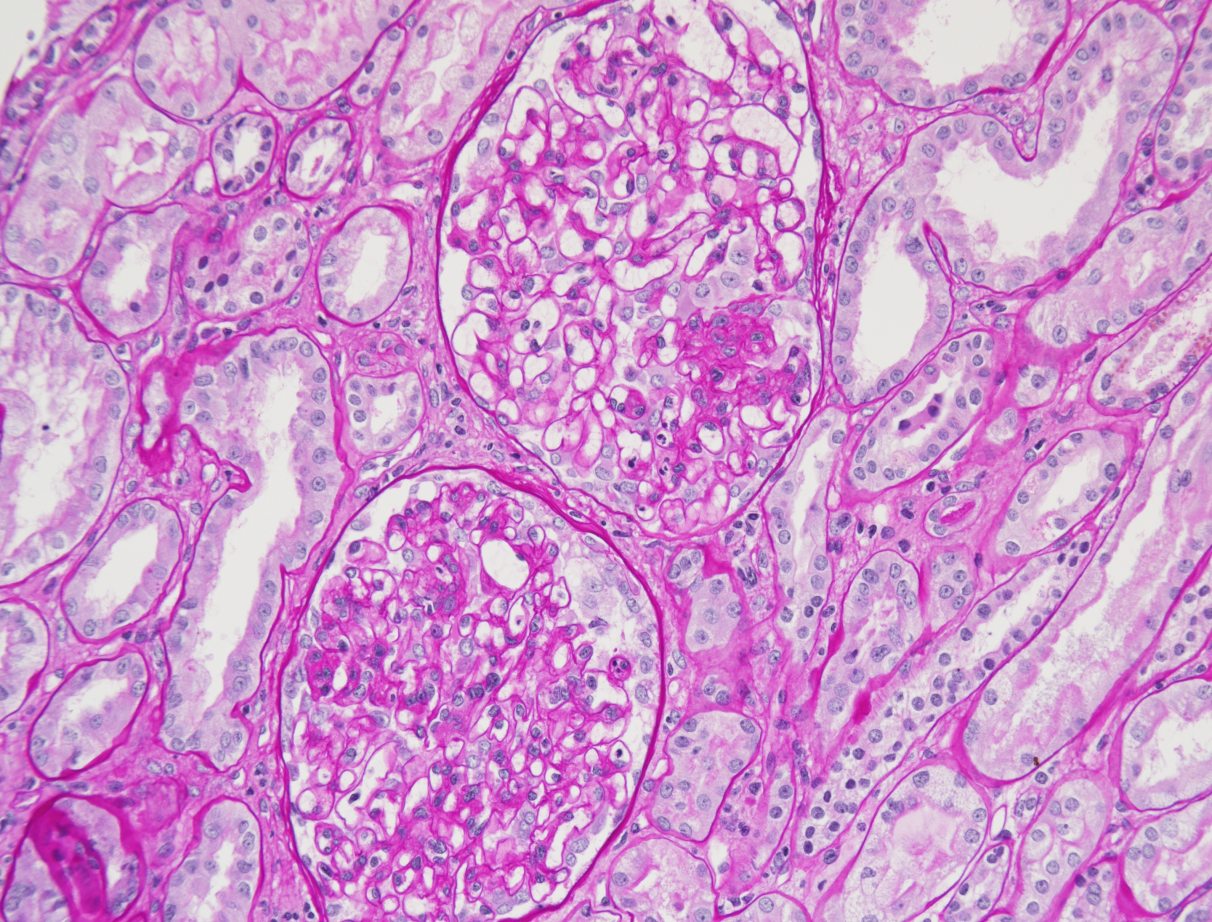
La malattia di Kikuchi-Fujimoto (KFD), o linfoadenite necrotizzante istiocitaria, è una malattia rara, con distribuzione mondiale ma è meglio conosciuta in Giappone e nell’Asia meridionale. La caratteristica più comune è la linfoadenopatia cervicale, accompagnata da dolorabilità o febbre alta, con sudorazione notturna, ma può anche essere asintomatica o con una gamma di sintomi molto ampia. La diagnosi è istopatologica, sulla biopsia escissionale. La malattia di Kikuchi-Fujmoto può imitare il linfoma ma anche la tubercolosi e alcune malattie autoimmuni, o essere associata a esse. È molto importante che la conosca anche il nefrologo per il suo possibile coinvolgimento renale. L’associazione con il LES è la più frequente ma non l’unica. Una diagnosi precoce di questa malattia può evitare accertamenti inutili e terapie aggressive.
Parole chiave: malattia di Kikuchi-Fujimoto, linfoadenite necrotizzante istiocitaria, coinvolgimento renale, biopsia escissionale, diagnosi differenziale


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}