

Abstract
L’acidosi metabolica è di frequente riscontro in clinica, in particolare nel paziente critico e/o con insufficienza renale. È sostenuta da meccanismi complessi, nella maggior parte dei casi individuabili attraverso l’anamnesi accurata, il corretto ragionamento diagnostico fisiopatologico, e la conoscenza di alcuni parametri di valutazione dell’equilibrio acido-base che, nella maggior parte dei casi, sono facilmente ottenibili al letto del malato. Su queste basi è possibile un rapido inquadramento, utile anche dal punto di vista pratico in condizioni di urgenza/emergenza, in due tipologie principali: le acidosi metaboliche ad elevato gap anionico e le acidosi metaboliche a gap anionico normale o ipercloremiche.
L’acidosi metabolica, soprattutto nella sua forma acuta, può compromettere in maniera significativa l’assetto emodinamico e metabolico dell’organismo, configurando situazioni di effettiva urgenza/emergenza. In tali condizioni cliniche l’approccio terapeutico non può quindi prescindere dalla rapida messa in atto di misure volte a risolvere problemi clinici concomitanti spesso alla base dello squilibrio stesso (ad esempio ottimizzazione emodinamica e fluidi in caso di shock, ventilazione meccanica in caso di insufficienza respiratoria concomitante, emodialisi per la rimozione di tossici etc.), in parallelo alla diagnostica differenziale. In caso di acidosi grave la somministrazione di agenti alcalinizzanti dovrà essere attentamente valutata sulla base dei possibili effetti collaterali, così come l’eventuale ricorso alla terapia sostitutiva della funzione renale.
Introduzione
L’acidosi metabolica è una alterazione dell’equilibrio acido–base caratterizzata da riduzione della bicarbonatemia, associata o meno ad acidemia (incremento della concentrazione idrogenionica, individuata in base alla riduzione dei valori di pH al di sotto di 7.36).
L’acidosi, sia in acuto che in cronico, è clinicamente rilevante in quanto si associa a significative alterazioni del metabolismo cellulare, contribuendo a modificare in senso negativo la prognosi del paziente, in termini di incremento sia della morbilità che della mortalità [1] (full text).
Questa rassegna ha lo scopo di illustrare i criteri di diagnosi, e i principali meccanismi patogenetici dell’acidosi metabolica, fornendo le basi per un approccio diagnostico pratico basato sul corretto ragionamento fisiopatologico. Verranno inoltre approfonditi i principi di trattamento di tale alterazione dell’equilibrio acido-base, sia in condizioni di emergenza/urgenza che in cronico, delineando i principi per il trattamento. Infine, saranno discusse in dettaglio le acidosi metaboliche a gap anionico normale (o ipercloremiche) e quelle a gap anionico elevato.
Aspetti epidemiologici e prognostici
Nonostante l’acidosi metabolica sia generalmente considerata di frequente riscontro, non sono numerosi gli studi che forniscono dati effettivi sull’epidemiologia e la prognosi di tale alterazione. Nella popolazione generale dei soggetti ospedalizzati l’acidosi metabolica è presente nell’1.9% dei soggetti, sebbene tale percentuale possa raggiungere valori ben più elevati (circa il 20%) in caso di coesistente riduzione della funzione renale [2] (full text). I dati della letteratura documentano valori di incidenza per l’acidosi metabolica compresi tra il 14% ed il 64% nei pazienti in terapia intensiva, soprattutto in quelli con insufficienza renale o con trauma maggiore [3] (full text). La forma più frequente è l’acidosi lattica (44%), seguita da altre forme di acidosi metabolica con gap anionico aumentato (37%), e da ultime sono le acidosi ipercloremiche (19% dei casi) [3] (full text). Circa l’8% dei pazienti può presentare un quadro di acidosi “grave” (pH < 7.20) [1] (full text). Nella maggior parte dei casi l’acidosi metabolica è secondaria ad altri problemi clinici (ipoperfusione sistemica o distrettuale, chetoacidosi, intossicazione da farmaci, etc), e può risolversi parallelamente al trattamento della causa sottostante. Studi fisiologici hanno tuttavia ipotizzato come valori di pH < 7.10 di possano di se stessi possa contribuire alla disfunzione d’organo, attraverso meccanismi quali l’aumento del lavoro dei muscoli respiratori, la ridotta diffusione di ossigeno ai tessuti, la riduzione della produzione di ATP e l’aggravamento della sepsi attraverso la modulazione della risposta immunitaria; inoltre l’acidosi potrebbe contribuire all’instabilità emodinamica deprimendo la contrattilità del ventricolo sinistro, facilitando l’insorgenza di aritmie e riducendo la sensibilità del letto vascolare ai vasopressori [1] (full text) [4] (full text) [5] (full text). La presenza di acidosi metabolica si associa ad un aumento dei tempi di degenza in terapia intensiva, oltre che della mortalità complessiva (fino al 57% nei pazienti con acidosi grave) [1] (full text) [4] (full text). Nel paziente critico, la persistenza di uno stato di acidosi grave a distanza di 24-48 ore dal ricovero si associa ad un elevato tasso di mortalità, indipendentemente dal valore di pH iniziale [1] (full text). Alcuni studi presentano un maggior tasso di mortalità per l’acidosi lattica e le acidosi ad elevato gap anionico (anion gap, AG) rispetto alle acidosi ipercloremiche, ma non è chiaro se ciò dipenda effettivamente da un peggioramento della prognosi legato in maniera specifica alle forme specifiche di acidosi opure alle comorbilità sottostanti [1] (full text) [3] (full text) [4] (full text) [6]. L’acidosi metabolica ipercloremica concomitante alla somministrazione di ingenti quantità di cristalloidi a base di cloro è un problema crescente nei pazienti critici in terapia intensiva, ed è stata associata a maggiore incidenza di insufficienza renale, prolungamento della dipendenza dalla ventilazione e dei tempi di ricovero [7] (full text). È stata recentemente segnalata un’elevata incidenza di acidosi tubulare renale anche nei pazienti critici in terapia intensiva (31%) [8] (full text), anche se il dato al momento non risulta confermato, e potrebbe essere gravato da alcuni errori metodologici. Nelle acidosi da intossicazioni esogene, di solito ad elevato AG, la relazione tra mortalità e pH sembra essere maggiormente documentata [9] [10] (full text). Anche nei pazienti con insufficienza renale cronica ricoverati in terapia intensiva l’acidosi metabolica si associa ad un peggioramento della prognosi [6] [11], ed è ipotizzabile che lo squilibrio non solo rappresenti un marker di gravità della condizione clinica sottostante, ma possa anche contribuire direttamente all’outcome negativo [12], attraverso i meccanismi sopracitati. L’acidosi metabolica costituisce inoltre una frequente complicanza nell’insufficienza renale cronica indipendentemente dall’eziologia di essa, con una prevalenza che aumenta progressivamente per valori di eGFR inferiori a 40 ml/min (15-39%) [13] (full text) [14] (full text). In cronico l’acidosi metabolica si associa ad una serie di problemi clinici quali aumentato riassorbimento osseo, riduzione della riserva respiratoria, esaurimento dei tamponi, riduzione dell’attività della Na/K ATPasi nei globuli rossi e nelle cellule miocardiche, con conseguente inotropismo negativo e scompenso cardiaco, sviluppo/aggravamento di insulino-resistenza ed ipertrigliceridemia, aumento dell’infiammazione, catabolismo muscolare e peggioramento dello stato nutrizionale [2] (full text). La maggior parte degli studi osservazionali ha mostrato una correlazione tra acidosi ed aumento della mortalità nei pazienti con CKD, sia in terapia conservativa che in dialisi cronica [15] (full text) [16] (full text) [17] (full text) [18] [19] [20] [21] (full text). Ridotti valori di bicarbonatemia si associano inoltre ad una più rapida progressione dell’insufficienza renale cronica nei pazienti in trattamento conservativo [22], ed a una più rapida riduzione del GFR rispetto alla popolazione generale anche nei pazienti senza insufficienza renale cronica [23]. Il meccanismo alla base di tale associazione non è del tutto definito, e la relazione potrebbe non essere causale, anche se almeno in parte un ruolo potrebbe essere attribuito alla risposta adattativa da parte dei nefroni residui. L’acidosi metabolica infatti promuove un aumento della produzione ed escrezione di ammonio, che si associa ad attivazione del complemento, del sistema renina-angiotensiona e ad aumento della produzione di endotelina-1, tutti fattori associati a stimolazione della fibrosi tubulo-interstiziale ed a danno renale progressivo [24] (full text) [25] (full text) [26] (full text).
Meccanismi di adattamento fisiologici alle variazioni del carico idrogenionico
Il mantenimento dell’omeostasi acido-basica consente di rispondere in maniera rapida ed efficiente alle variazioni del carico idrogenionico, evitando in tale modo l’accumulo di acidi o di basi nell’organismo. In condizioni normali il più importante fattore esterno che può influenzare l’omeostasi acido-base è rappresentato dalla dieta. L’effetto della dieta sull’equilibrio acido-base dipende dal bilancio netto fra acidi, basi ed i loro precursori in essa contenuti. Il bilancio netto giornaliero tra acidi e basi della dieta di un soggetto non vegetariano è positivo per circa 70 mEq di H+ (1 mEq/Kg/die), ed in condizioni fisiologiche il mantenimento dell’omeostasi avviene attraverso l’eliminazione di tale eccesso di H+ da parte dell’organismo. I meccanismi fisiologici di adattamento e compenso ad un carico di H+ quale quello giornaliero dovuto alla dieta, prevedono tre ordini di risposta: i sistemi tampone, l’adattamento respiratorio ed il compenso (in realtà correzione) da parte del rene.
A. I sistemi tampone
I sistemi tampone rappresentano la prima linea di difesa dell’omeostasi acido-base, essendo in grado di ridurre in tempi brevi (minuti/ore) l’impatto sul pH di eventuali variazioni – in aumento o in diminuzione – del bilancio idrogenionico [27] (full text). Un tampone è formato da un acido debole e dalla sua base coniugata. La capacità di minimizzare le variazioni del pH, ovvero della concentrazione idrogenionica, dipendono dalle concentrazioni relative delle due specie ioniche, e dal pKa specifico del sistema tampone. Un sistema tampone è massimamente efficace quando le concentrazioni dell’acido e della base coniugata si equivalgono. Ciò si verifica quando il pH della soluzione (o dell’organismo) in cui si trova ad essere il tampone corrisponde al pKa del sistema tampone stesso (cioè il valore di pH al quale le componenti acida e basica del sistema tampone sono presenti ciascuna per il 50%). Nell’organismo sono presenti diversi sistemi tampone, localizzati nel compartimento extracellulare, all’interno delle cellule, nei globuli rossi e nel tessuto osseo. Tali sistemi si trovano in equilibrio tra loro secondo il principio isoidrico, per cui lo studio del tampone prevalente sarà informativo anche sullo stato degli altri tamponi, ed in generale sull’equilibrio acido-base dell’organismo. Questo è il motivo per cui la valutazione dell’equilibrio acido-base può essere ottenuta dall’analisi di un solo sistema tampone, quello principale, che nel plasma è rappresentato dal sistema acido carbonico/bicarbonato [28] [29]. Quest’ultimo, oltre a svolgere un ruolo centrale nel mantenimento dell’omeostasi acido-base, presenta componenti facilmente misurabili sul sangue arterioso attraverso un esame semplice e sempre disponibile – l’emogasanalisi – che fornisce gli elementi, attraverso l’equazione di Henderson-Hasselbach, per il più importante metodo di interpretazione dello stato acido-base sistemico, ed indirettamente degli altri sistemi tampone dell’organismo. Rispetto agli altri tamponi, il sistema acido carbonico/bicarbonato presenta delle peculiarità. Infatti essendo costituito da un “sistema aperto” in cui la CO2 che si forma dall’acido carbonico può essere rapidamente allontanata dall’organismo attraverso l’incremento della ventilazione alveolare, risulta relativamente indipendente in termini di effetto tampone dal pH della soluzione in cui si trova ad operare.
[CO2] [H2O] ↔ [H2CO3] ↔ [H+] [HCO3–] pH= 6.10 + log[pCO2] /[HCO3–]Altri tamponi quantitativamente meno rappresentati nel compartimento extracellulare sono costituiti dai fosfati inorganici e dai gruppi istidinici dell’albumina e delle globuline plasmatiche. Una percentuale variabile del carico acido viene tamponato a livello intracellulare, in particolar modo nel muscolo scheletrico [30] [31] (full text) e nell’osso [30]. L’ingresso di H+ nelle cellule determina la fuoriuscita di K+ al fine di mantenere l’elettroneutralità. Questo effetto è maggiore nelle forme di acidosi metabolica dovute ad un eccesso di acidi non organici, come in corso di diarrea o nell’insufficienza renale, in cui non è presente un anione coniugato. In corso di acidosi l’idrossiapatite che costituisce il tessuto osseo, agisce come donatore di OH–, tamponando l’eccesso di H+ e liberando fosfati e bicarbonato. Mentre in passato si riteneva che questo effetto dipendesse unicamente da una dissoluzione puramente fisico-chimica dalla matrice ossea, alcuni studi su modelli animali e nell’uomo hanno dimostrato che anche modeste variazioni del pH (< 0.1 unità) sono in grado di attivare significativamente ed in maniera duratura gli osteoclasti, inibendo al contempo la deposizione di matrice ossea da parte degli osteoblasti. Questo effetto è mediato direttamente da sensori di pH associati a proteine G espressi sulla membrana sia degli osteoclasti che degli osteoblasti [30]. Un effetto analogo sarebbe ascrivibile all’ipossia tissutale [30]. Nel muscolo scheletrico avviene invece la maggior parte del tamponamento effettuato dal sistema bicarbonato, la cui efficienza può essere stimata dai livelli di PCO2 su sangue venoso refluo da un distretto muscolare (ad esempio brachiale o femorale), che a sua volta riflette la PCO2 capillare dei muscoli scheletrici stessi [28]. In condizioni di normale perfusione muscolare, la PCO2 venosa brachiale è circa 46 mmHg, mentre la PaCO2 (arteriosa) è attorno a 40 mmHg. Tale gradiente assicura una costante diffusione di CO2verso il polmone, e poiché in tale modo vengono garantiti bassi livelli di PCO2 venosa, la maggior parte del carico di H+ potrà essere tamponato dal bicarbonato (Figura 1). In presenza di ipoperfusione, la differenza tra PCO2 venosa e PaCO2 aumenta significativamente (> 10 mmHg), e l’aumentato carico di H+ circolanti verrà tamponato in misura sempre maggiore dai tamponi non bicarbonato, ovvero dalle proteine presenti nel citosol delle cellule [31] (full text)(Figura 1). La conseguenza più immediata è che il legame degli H+ alle proteine intracellulari ne altera la struttura, la carica e la funzione [31] (full text), con effetti rilevanti dal punto di vista clinico, in particolare nei parenchimi nobili come cuore e sistema nervoso centrale. A tale proposito, è opportuno sottolineare come il punto di vista tradizionale “protone-centrato”, focalizzato prevalentemente sulla concentrazione degli ioni idrogeno nel sangue arterioso, non consideri l’eventuale costo metabolico e funzionale derivante dal tamponamento intracellulare degli H+ da parte dei tamponi non bicarbonato, con le possibili conseguenze strutturali e funzionali di esso sulle proteine contrattili, enzimatiche etc. È quindi possibile che un’interpretazione più “proteino-centrata” degli effetti fisiopatologici del tamponamento intracellulare possa offrire una migliore comprensione degli effetti dell’acidosi, oltre ad avere importanti implicazioni terapeutiche. Il ruolo dei sistemi tampone non sarebbe più quindi semplicemente quello di abbassare la concentrazione degli idrogenioni – e sotto questo punto di vista sarebbe indifferente quale sistema tampone venga utilizzato – ma soprattutto quello di minimizzare il legame degli H+ alle proteine nelle cellule degli organi vitali (cervello, cuore). Sotto questo punto di vista il sistema bicarbonato è effettivamente il tampone più efficace, ma solo se il prodotto del tamponamento (CO2) può essere rapidamente rimosso, evitando l’accumulo locale di H+, e l’inevitabile ricorso ad altri sistemi tampone (proteine), meno spendibili nell’economia generale dell’organismo [31] (full text).
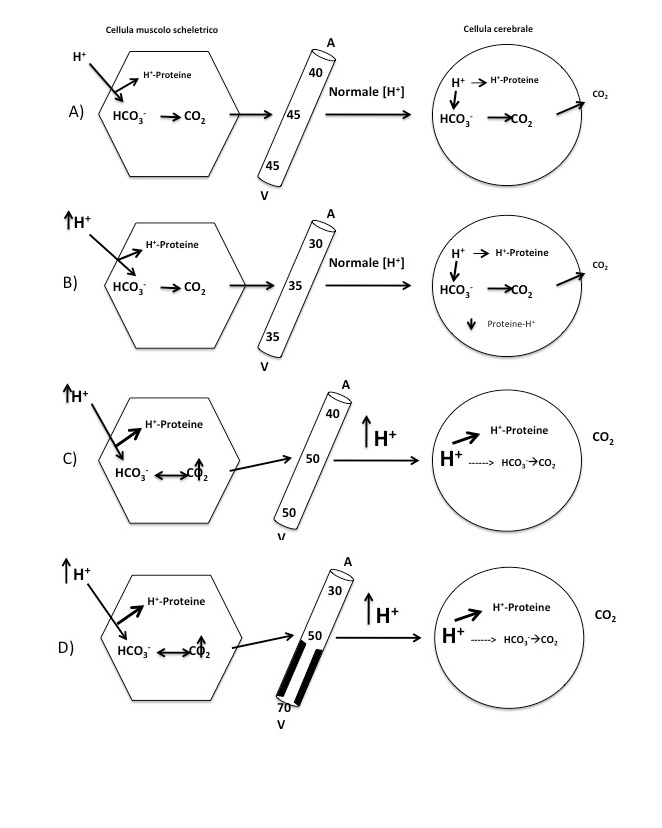{kind=link}
B. Adattamento respiratorio
L’adattamento respiratorio (a volte impropriamente indicato come “compenso”) all’acidosi metabolica, in base all’equazione di Henderson-Halsselbalch consiste nella riduzione della PaCO2 attraverso l’aumento della ventilazione alveolare. Il processo inizia immediatamente all’instaurarsi dell’acidosi, ma raggiunge il suo massimo solo dopo 12-24 ore. Tale “ritardo” nella risposta è dovuto al differente comportamento dei chemiocettori centrali e periferici. Infatti, nella prima ora dall’instaurarsi dell’acidosi il processo di compenso è mediato principalmente dai chemiocettori localizzati nei corpi carotidei, che per primi risentono di una variazione del pH plasmatico. Come risposta, essi stimolano i centri respiratori bulbari, determinando un incremento della ventilazione, ed una conseguente riduzione della PaCO2. Tuttavia a seguito della maggiore permeabilità della barriera ematoencefalica alla CO2 rispetto al HCO3–, per la stessa equazione di Henderson-Halsselbalch, il pH liquorale risentirà più rapidamente della caduta della CO2 indotta dall’iperventilazione, rispetto alla riduzione del bicarbonato, determinando un paradossale aumento del pH liquorale, che inizialmente limita la risposta ventilatoria. Se però l’acidemia persiste, la progressiva redistribuzione degli ioni attraverso la barriera emato-encefalica determinerà una riduzione sia della bicarbonatemia liquorale che del pH, con conseguente stimolo ventilatorio centrale che andrà a sommarsi a quello periferico dei chemiocettori carotidei [32] [33]. L’eliminazione di H+ come acido “volatile” attraverso l’adattamento respiratorio costituisce un altro meccanismo di risparmio dei tamponi proteici intracellulari. L’entità del’adattamento respiratorio è quantificabile in una riduzione della PaCO2di 0.8-1.2 mmHg per ogni mEq/L di riduzione della bicarbonatemia (ovvero del ΔHCO3–) [33]. Una differenza tra i valori di PaCO2 osservati ed attesi in relazione all’adattamento respiratorio prevedibile suggerisce la presenza di un disturbo misto. L’adattamento respiratorio, per quanto efficace, risulta limitato nel tempo. Infatti, la riduzione della PaCO2 determina progressivamente una parallela riduzione del riassorbimento tubulare di bicarbonato, che fa sì che dopo un certo lasso di tempo il pH ritorni ai valori precedenti l’inizio dell’adattamento respiratorio.
C. Compenso (correzione) renale
Il rene può attuare un vero e proprio “compenso” all’acidosi metabolica, in quanto è l’unico organo in grado di correggere attivamente i valori di bicarbonato, modificandone la rigenerazione o l’eliminazione in base delle necessità dell’organismo. Ovviamente l’organo non deve essere la causa primitiva dell’acidosi metabolica, cioè non devono essere presenti patologie renali che alterino primitivamente i processi di acidificazione (o di eliminazione di basi). In condizioni fisiologiche (e di dieta standard, che nella maggior parte dei casi è acidificante), il rene contribuisce al mantenimento dell’equilibrio acido-base attraverso due processi:
- Il riassorbimento di tutto il bicarbonato filtrato a livello glomerulare
- L’eliminazione di H+ (attraverso l’acidità titolabile e l’ammoniuria), alla quale è obbligatoriamente accoppiata la rigenerazione del bicarbonato eventualmente consumato
Rovesciando il ragionamento (ad es. nel caso di una dieta alcalinizzante, come quelle vegetariane), è ovvio che il rene – allo stesso modo – sarà anche in grado di eliminare un eccesso di basi, sotto forma di bicarbonato.
Il bicarbonato attraversa liberamente la barriera glomerulare. In condizioni normali, vengono filtrati circa 4500 mEq di bicarbonato nelle 24 ore (GFR 180 L/die x 24-25 mEq/L), che devono essere interamente riassorbiti per mantenere l’omeostasi [33]. Circa il 90% del bicarbonato filtrato è riassorbito nel tubulo prossimale, e solo il restante 10% nei segmenti distali, nel tratto spesso ascendente dell’ansa di Henle e nei dotti collettori della midollare esterna [33] [34] (full text). A livello del tubulo prossimale il riassorbimento di HCO3– avviene quasi interamente (90%) con meccanismo indiretto mediante secrezione di H+ nel lume tubulare attraverso l’isoforma NHE3 dello scambiatore Na+/H+ (Figura 2). L’H+ secreto si combina con l’HCO3– filtrato formando acido carbonico, che a sua volta viene dissociato a CO2 ed H2O dall’anidrasi carbonica IV (AC IV) presente sull’orletto a spazzola delle cellule tubulari. La CO2 e l’H2O entrano nelle cellule epiteliali prossimali dove viene formato nuovamente HCO3– per azione dell’anidrasi carbonica II (AC II), con produzione di protoni che vengono nuovamente secreti nel lume [33] [34] (full text). Il bicarbonato così formatosi fuoriesce dalle cellule verso il compartimento interstiziale, per effetto del cotrasportatore basolaterale Na+/HCO3–, (SLC4A4) (Figura 2). La presenza dell’anidrasi carbonica permette di riassorbire la maggior parte del bicarbonato filtrato senza determinare acidificazione delle urine, che limiterebbe la secrezione di idrogenioni: le cellule del tubulo prossimale non sono infatti in grado di creare un forte gradiente di H+ tra l’intracellulare ed il lume tubulare. Il gradiente elettrochimico che garantisce questi spostamenti è fornito dal riassorbimento del sodio a livello della membrana basolaterale, per azione della pompa 3Na+/2K+-ATPasi, che garantisce il mantenimento di basse concentrazioni intracellulari di sodio e di un gradiente intracellulare negativo. Risulta chiaro quindi che nel tratto prossimale del nefrone il riassorbimento del bicarbonato è direttamente associato a quello del sodio. Nel tratto distale invece, la maggior parte del sodio filtrato è già stato riassorbito (in particolar modo lo sarà negli stati di ipovolemia), ed il pH urinario si riduce progressivamente, incrementando il gradiente contro cui gli idrogenioni devono essere secreti. Per questo motivo l’acidificazione di questo tratto deve essere mediata da un trasporto attivo, basato sulle H+-ATPasi e H+/K+-ATPasi presenti nelle cellule intercalate tipo α del dotto collettore (Figura 3 parte a). La regolazione delle H+-ATPasi avviene attraverso l’incorporazione di vescicole nella membrana citoplasmatica, analogamente alle aquaporine [33]. I maggiori determinanti del riassorbimento del bicarbonato includono la concentrazione di HCO3– e il pH nel lume tubulare, la velocità di flusso luminale, la pressione parziale peritubulare di CO2 e le concentrazioni luminali e peritubulari di angiotensina II [35]. Il secondo processo consiste nell’eliminazione del carico giornaliero di H+prodotti dal metabolismo degli acidi fissi introdotti con la dieta, con conseguente rigenerazione dei bicarbonati consumati. Solo una minima quota di H+ è escreta come ione libero. Infatti al pH urinario minimo raggiungibile (circa 4.5), la quota libera di H+ rappresenta meno dell’1% della quantità totale da eliminare, mentre la maggior parte della quota acida è escreta mediante coniugazione con tamponi urinari come il fosfato (pKa 6.80) (Figura 3 parte b), la creatinina (pKa 4.97), l’acido urico (pKa 5.75) e soprattutto l’ammoniaca (pKa 9) (Figura 3 parte c) [33]. Se si considerano le caratteristiche dei sistemi tamponi elencati in precedenza, teoricamente il tampone più efficiente al pH urinario dovrebbe essere costituito dal fosfato. Tuttavia l’efficacia di questo tampone dipende direttamente dal carico di fosfato filtrato, e nonostante sia dimostrato un lieve incremento della fosfaturia indotta dall’acidosi, il potere tampone tende ad esaurirsi rapidamente allorché la pre-urina viene acidificata lungo il tubulo. Quindi, il sistema tampone del fosfato non è in grado di far fronte quantitativamente efficace ad un incremento significativo del carico acido netto. Viceversa, nonostante il sistema NH3/NH4+ presenti un pKa più sfavorevole al pH urinario abituale del nefrone distale, di fatto viene a costituire il principale sistema di eliminazione del carico acido netto. Ciò è dovuto fondamentalmente a due fattori:
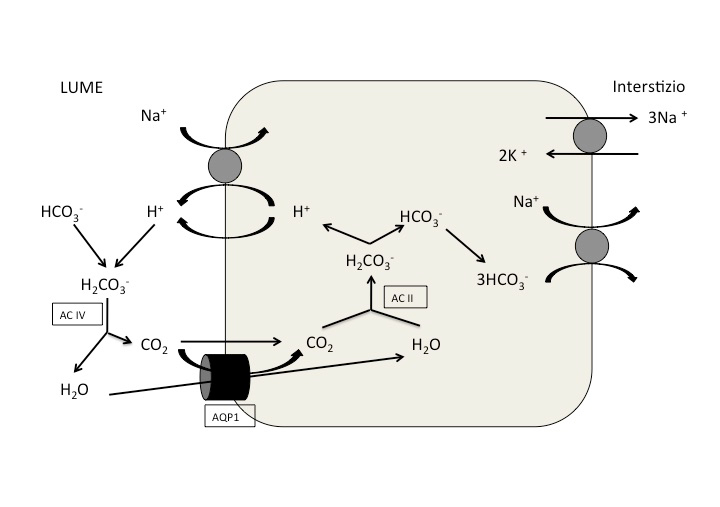{kind=link}
{kind=link}
- La possibilità di aumentare la produzione di NH3 a partire dal metabolismo della glutamina che all’interno delle cellule del tubulo prossimale è modulabile (aumentata disponibilità indotta dall’acidosi)
- La differente permeabilità dei diversi tratti del tubulo allo ione ammonio, che permettono di formare un “serbatoio” di NH3 nella midollare, che va a rifornire il sistema tampone man mano che viene consumato
La produzione di ione ammonio avviene prevalentemente nelle cellule del tubulo prossimale a partire dal catabolismo della glutamina (Figura 4). Tale processo viene attivato dal carico cronico di H+, che stimola l’ingresso di glutamina nei mitocondri delle cellule tubulari ed il catabolismo di essa, mediante la glutaminasi e la glutammato-deidrogenasi, fino a glutammato ed α-ketoglutarato, con produzione netta per ogni molecola di glutamina di 2 ioni ammonio (escreti nelle urine) e di 2 ioni bicarbonato (immessi in circolo) [33]. Lo ione ammonio viene secreto nel lume del tubulo prossimale e riassorbito nel tratto spesso del tratto ascendente dell’ansa di Henle in sostituzione del potassio a livello del cotrasportatore Na+/K+/2Cl– Il conseguente incremento della potassiuria, legato alla competizione tra K+ e NH4+ per il cotrasportatore, fornisce anche un meccanismo di difesa dall’iperpotassiemia in corso di acidosi metabolica. L’NH3 accumulato nella midollare può progressivamente diffondere attraverso la membrana del tubulo collettore, tamponando gli idrogenioni nel lume tubulare e riducendo così la caduta del pH urinario nelle porzioni distali del nefrone (Figura 4). L’escrezione di ammonio assicura normalmente l’eliminazione di 30-40 mEq/die di H+, ai quali si sommano circa 20-30 mEq/die legati agli altri tamponi, soprattutto fosfati [33]. In corso di acidosi metabolica da cause extrarenali il rene può però incrementare notevolmente l’escrezione di H+ (fino ad oltre 200 mmol/die), aumentando la produzione e l’escrezione di NH3. Ne deriva quindi che la risposta adattativa renale all’acidosi metabolica consiste principalmente in un aumento dell’escrezione acida netta sotto forma di NH4+ (ovviamente nei casi in cui il rene stesso non sia la causa dell’acidosi metabolica). La quantificazione diretta o indiretta dell’ammoniuria assume quindi un significato fondamentale in termini di diagnosi differenziale, consentendo di distinguere le forme di acidosi metabolica da cause renali o extrarenali [28]. Il processo di acidificazione urinaria è fondamentale in quanto sia l’eliminazione di acidità titolabile che la formazione di ammonio sono pH-dipendenti. Infatti, i diversi sistemi tampone presenti nel filtrato glomerulare giocano un ruolo più o meno rilevante in base al pH presente nei vari tratti del tubulo. Nel tratto prossimale, che presenta un pH relativamente alcalino (6.80), i tamponi prevalenti sono il bicarbonato, che viene in gran parte riassorbito attraverso l’anidrasi carbonica, ed il fosfato. Questo tratto è inoltre il principale sito di secrezione di NH4+. Nel tratto distale invece, a livello del dotto collettore, dove il pH scende ai valori alla massima acidificazione, gli H+ secreti si combineranno esclusivamente con NH3, visto che tutto il bicarbonato è già stato riassorbito; allo stesso modo tutto il fosfato sarà già stato tamponato nei segmenti precedenti del nefrone, che presentano valori di pH di 6.8 rispetto al pH di questo tratto che scende al di sotto di 5.8) [33]. L’eliminazione di H+ e di conseguenza il pH plasmatico, viene inoltre influenzata da una serie di altri fattori quali la volemia efficace, l’aldosterone, la potassiemia ed il PTH. Il riassorbimento di bicarbonati è infatti inversamente proporzionale al volume circolante poiché, come visto in precedenza, il riassorbimento prossimale del bicarbonato avviene in gran parte in associazione al sodio. In condizioni di euvolemia il riassorbimento di bicarbonato raggiunge un plateau per concentrazioni plasmatiche fisiologiche (circa 26 mEq/L). In caso di ipovolemia invece, si attivano i meccanismi sodio-ritentivi, che tendono a mantenere il riassorbimento del sodio anche a spese di un accumulo di bicarbonati. A ciò contribuiscono fattori differenti, quali la riduzione del GFR, l’attivazione del SRAA, l’ipocloremia e l’eventuale contemporanea ipokaliemia [28] [33].
{kind=link}
Patogenesi e classificazione delle acidosi metaboliche
Il riconoscimento del meccanismo che ha determinato uno stato acidosico è fondamentale ai fini di una corretta terapia. Il calcolo di AG, con le dovute limitazioni interpretative, è fondamentale nella diagnosi differenziale dell’acidosi metabolica. Fisiologicamente la somma algebrica degli ioni misurabili nel plasma non corrisponde a zero, ma si attesta attorno a 8-12 mEq/L, e tale variabilità dipende dai valori di normalità di ciascun laboratorio e dell’individuo. Tale differenza è dovuta di solito alle cariche elettriche negative presenti sulla superficie delle proteine plasmatiche, in primo luogo l’albumina. Le proteine plasmatiche quindi, anche se nella pratica clinica non vengono misurate in mEq/L, bensì in unità di massa per volume (g/dL), rientrano nel bilancio ionico atto a mantenere l’elettroneutralità (Figura 5). Tenendo presente questo concetto, il valore normale di AG risulterà essere influenzato in larga misura dai valori di albuminemia, e quindi dovrà essere corretto per variazioni della concentrazione plasmatica di tale proteina (il gap anionico si riduce di 2,5 mEq/L per ogni g/dL di riduzione dei valori di albuminemia) [28]. La perdita di bicarbonati nelle urine o nelle feci si associa a obbligatoria ritenzione di Cl–, l’unico altro anione disponibile in rilevanti quantità, al fine di mantenere l’elettroneutralità e la volemia: tale effetto, se non sono presenti altri anioni, equivale chimicamente alla somministrazione di HCl in rapporto equimolare con la perdita di bicarbonati. In questo caso AG non si modificherà in quanto, per ogni molecola di bicarbonato che viene persa, l’elettroneutralità è mantenuta da un corrispondente anione cloro. Si verrà in tale modo a definire una condizione di acidosi metabolica a AG normale, o ipercloremica. Quando invece l’acido che viene aggiunto è diverso dal HCl (cioè non vi è perdita di bicarbonati) si verificherà l’aggiunta di un anione non misurato, in un rapporto teorico di 1:1 rispetto alla riduzione del bicarbonato. L’’effetto sarà quello di un parallelo aumento di AG. Se l’acidosi dipende dall’accumulo di acidi diversi da HCl (es acido lattico o chetoacidi) che si associano all’accumulo di anioni non misurati, idealmente in un sistema monocompartimentale si avrà un aumento di AG dovuto a riduzione dei valori di bicarbonato corrispondente all’accumulo della base coniugata dell’acido (l’anione non misurato). Di conseguenza il rapporto tra il Δ aumento AG e Δ riduzione HCO3– (il cosiddetto delta/delta) sarà circa 1. Spesso tuttavia tale rapporto si discosta da 1 a causa di alcuni fattori che devono essere tenuti in considerazione [31] (full text):
{kind=link}
- La presenza del tamponamento intracellulare.
- L’eliminazione renale della base coniugata in associazione ad un catione diverso dall’ammonio, generalmente Na+ o K+, cosa che tende a normalizzare AG. Questo è ciò che avviene tipicamente nelle fasi di riespansione volemica in corso di chetoacidosi diabetica.
- La presenza di disturbi misti (es. acidosi ipercloremica +acidosi lattica in corso di diarrea profusa con ipovolemia grave, acidosi lattica + alcalosi metabolica in caso di vomito protratto con ipovolemia grave).
Le limitazioni all’impiego di AG derivano anche dalla necessità di stimare il valore abituale di AG del paziente allo stato di equilibrio. Infatti altri fattori quali calcemia, magnesemia oppure variazioni nella concentrazione delle proteine plasmatiche o dei fosfati dovute all’emoconcentrazione o alla presenza di paraproteine, possono modificare AG anche indipendentemente da variazioni dell’equilibrio acido-base. L’emoconcentrazione costituisce una condizione da tenere in particolare considerazione in quanto l’incremento delle valenze anioniche dell’albumina dovute alla contrazione del volume extracellulare (ECF) può determinare apparente aumento di AG, suggerendo erroneamente un accumulo di acidi. Al fine di un più corretto inquadramento del disturbo è utile quindi considerare anche l’albuminemia, l’ematocrito e PvCO2 come parametri indiretti dell’ECF [28] [36]. In specifici contesti (es. intossicazioni da alcooli e glicoli), l’inquadramento diagnostico può essere completato dal gap osmolale plasmatico (OG), che permette di sospettare la presenza di sostanze normalmente non misurate, che però costituiscono osmoli efficaci (es. metanolo). L’OG è dato dalla differenza tra l’osmolalità misurata e quella calcolata.
OG = Posm – [2Na+p+glicemia/18+BUN/2,8]
Alternativamente all’approccio tradizionale centrato sul bicarbonato e sull’equazione di Henderson-Hasselbalch, è opportuno citare l’approccio di Stewart basato sulla “strong ion difference, SID” [27] (full text). Questo approccio, discostandosi dalla tradizionale definizione di Brönsted-Lowry, definisce come acido come uno ione in grado di spostare l’equilibrio di dissociazione dell’acqua verso gli H+, e come base verso gli OH–. In questa visione, la bicarbonatemia non costituisce più un determinante del pH, ma una variabile dipendente. I determinanti dell’equilibrio acido base verrebbero quindi ad essere costituiti esclusivamente dalla SID, dalla concentrazione plasmatica degli acidi deboli quali albumina e fosfati, che costituiscono i tamponi non volatili, e che in assenza di altri anioni non misurati corrispondono ad AG, e dalla PaCO2. Il SID corrisponde alla differenza tra gli ioni presenti nel plasma. Il SIDapp, ovvero il SID apparente, è dato dalla differenza degli ioni misurabili (Ca+, Mg+, K–, Cl–, lattato), e corrisponde a circa 40mEq. L’approccio di Stewart definisce sei alterazioni dell’equilibrio acido base (acidosi respiratoria, alcalosi respiratoria, acidosi da ioni forti, alcalosi da ioni forti, acidosi da tamponi non volatili, alcalosi da tamponi non volatili). In funzione del SIDapp il pH aumenta all’aumentare della differenza tra gli ioni forti e diminuisce col diminuire di tale differenza. Questo approccio spiegherebbe l’acidosi da diluizione che si osserva nei pazienti cui sono state somministrate grandi quantità di cristalloidi, in particolar modo soluzione fisiologica allo 0.9%, sulla base una ridotta differenza tra gli ioni Na+ e Cl–. Poiché il SID è funzione del pH e dei tamponi non volatili, può essere calcolato da questi (SID effettivo). La differenza tra il SID effettivo ed il SID apparente costituisce lo strong ion gap, che normalmente è zero, ma in caso di aggiunta di acidi organici endogeni o esogeni aumenta. Questo approccio seppure apparentemente più preciso e completo dal punto di vista chimico-fisico, risulta gravato da una maggiore complessità di calcolo, che richiede un’equazione polinomiale, e di fatto non risulta più efficace sul piano diagnostico e prognostico rispetto all’approccio tradizionale [37] (full text). Infatti alcuni studi hanno dimostrato che, qualora si corregga il gap anionico per l’albuminemia, nella maggioranza dei quadri clinici i due approcci sono sovrapponibili nell’inquadrare il disturbo acido-base sottostante [37] (full text) [38] (full text) [39] [40] [41] [42] [43] (full text). Per questi motivi in questa rassegna verrà fatto esclusivamente riferimento all’approccio tradizionale, che classifica le acidosi metaboliche da un punto di vista fisiopatologico. In quest’ottica l’acidosi metabolica può essere causata fondamentalmente da due meccanismi: perdita diretta di bicarbonati effettivi o potenziali (acidosi metabolica a normale gap anionico o ipercloremica), oppure aggiunta ai fluidi corporei di H+ derivanti da aumentata produzione/accumulo di acidi esogeni o endogeni, che come visto in precedenza determinano un consumo dei sistemi tampone e parallelo aumento di AG (acidosi metabolica a gap anionico aumentato) (Figura 6). Per ciascuna delle principali forme verranno descritti i più importanti esami che costituiscono la base per la semeiotica funzionale e la diagnosi differenziale.
{kind=link}
Acidosi metaboliche a gap anionico normale o acidosi ipercloremiche
Come già illustrato in precedenza la perdita di bicarbonati sotto forma di sali di Na+ o di K+, si associa a ritenzione di Cl– e determina acidosi metabolica ipercloremica o a normale AG.
La perdita di bicarbonati può avvenire a due livelli:
- A livello del tratto gastroenterico
- A livello renale.
A. Perdite Gastrointestinali
La più comune causa di perdita intestinale di bicarbonati è la diarrea acuta o cronica, ma più in generale tutte le secrezioni intestinali al di sotto del ligamento di Treitz contengono elevate quantità di bicarbonato (Tabella 1). A livello del colon sono secreti bicarbonati (almeno 30 mEq/L) per effetto dello scambiatore HCO3–/Cl–, ed inoltre può verificarsi la perdita di bicarbonati “potenziali” prodotti dalla fermentazione batterica (es butirrato, propionato, lattato) [44] (full text). La concentrazione relativamente bassa di bicarbonati nelle feci rende il pH fecale, che spesso è acido in corso di diarrea, un parametro poco attendibile per valutare l’effettiva perdita di basi. Una maggiore correlazione con la perdita di bicarbonati effettivi e potenziali in corso di diarrea può essere ottenuto dalla differenza tra il sodio, il potassio ed il cloro fecale, che corrisponde grossolanamente alla perdita di bicarbonati e di anioni organici nelle feci [44] (full text) [33]. In aggiunta alla perdita di basi dal tratto gastroenterico, in corso di diarrea è opportuno considerare altri fattori che possono determinare interferenze nell’interpretazione dei parametri sull’equilibrio acido base. In particolare, l’eventuale ipovolemia associata può da un lato, condizionare una sottostima dell’effettiva perdita di bicarbonati per emoconcentrazione, e dall’altro può determinare un deficit della capacità di acidificazione tubulare. Questa avviene a seguito della contrazione del GFR ed all’aumentato riassorbimento prossimale di sodio e cloro, che riduce la capacità di acidificazione nel tratto distale del tubulo e la produzione/escrezione di NH4+. La presenza di acidosi ed emoconcentrazione in corso di diarrea possono inoltre nascondere uno stato di deplezione di potassio, che può emergere al momento della correzione [28] [33]. Altre cause di acidosi metabolica da perdite intestinali di bicarbonato possono essere rappresentate da fistole pancreatiche e biliari, da drenaggi digiunali o pancreatici, dal drenaggio urinario dei succhi pancreatici in corso di trapianto combinato rene-pancreas [45], dalla presenza di uterosigmoidostomie o di neovescica ileale [33] [45]. Infine, i pazienti in terapia con colestiramina, magnesio o calcio possono sviluppare acidosi metabolica per effetto chelante di queste sostanze sul bicarbonato [45].
B. Perdite renali
La perdita diretta di bicarbonati per mancato riassorbimento tubulare, o indiretta, per ritenzione di H+ o ridotta produzione di NH4+, configura le acidosi tubulari renali (ATR). Le ATR sono alterazioni dell’equilibrio acido-base su base congenita, o più frequentemente acquisita, caratterizzate da compromissione del processo di acidificazione urinaria sproporzionata al grado di riduzione della funzione renale. È infatti noto come sia necessaria una marcata riduzione del GFR (al di sotto di 30-40 ml/min) perché si verifichi la comparsa di acidosi metabolica secondaria a riduzione della massa nefronica, mentre di solito nelle ATR la funzione renale è conservata o solo lievemente compromessa. Classicamente vengono individuate tre tipologie di ATR (tipo I, II e IV), più una forma (ATR III) non riconosciuta da tutti gli autori e che presenterebbe aspetti comuni sia all’ATR tipo I che a quella tipo II (Tabella 2). Tuttavia, l’utilizzazione di una classificazione mirata agli aspetti fisiopatologici risulta sicuramente più vantaggiosa, sia in termini di definizione dei meccanismi sottostanti, che per la diagnosi differenziale [28](Tabella 3). I principali test della semiotica funzionale delle acidosi metaboliche sono descritti in Tab. 4. Si distinguono forme di ATR prossimali, caratterizzate principalmente da specifica compromissione del riassorbimento di bicarbonato a livello del tubulo prossimale (ATR prossimale o tipo II), e forme di ATR distali, nelle quali la compromissione funzionale della capacità di acidificazione a livello della porzione distale del nefrone (tubulo distale e/o collettori) si riflette in una ridotta o assente escrezione acida netta. Le forme distali a loro volta possono essere distinte in forme ipopotassiemiche (ATR tipo I “classica”) e in forme iperpotassiemiche, ulteriormente suddivisibili in forme da alterazione generalizzata del nefrone distale (ATR tipo I “iperpotassiemica”), e in forme secondarie a ipoaldosteronismo/pseudoipoaldosteronismo, che corrispondono al tipo IV [45] [46] [47] [48] (full text). I meccanismi fisiopatologici e le corrispondenze tra le diverse classificazioni sono illustrati in Tab. 3. L’aspetto fisiopatologico comune in tutte le forme di ATR – ed elemento centrale nella diagnosi – è comunque una eliminazione di ammonio (NH4+) inadeguata rispetto alla presenza ed all’entità dell’acidosi metabolica. In Figura 7 è descritto un algoritmo per la diagnosi differenziale dell’acidosi metabolica a gap anionico normale (o ipercloremica) e delle ATR.
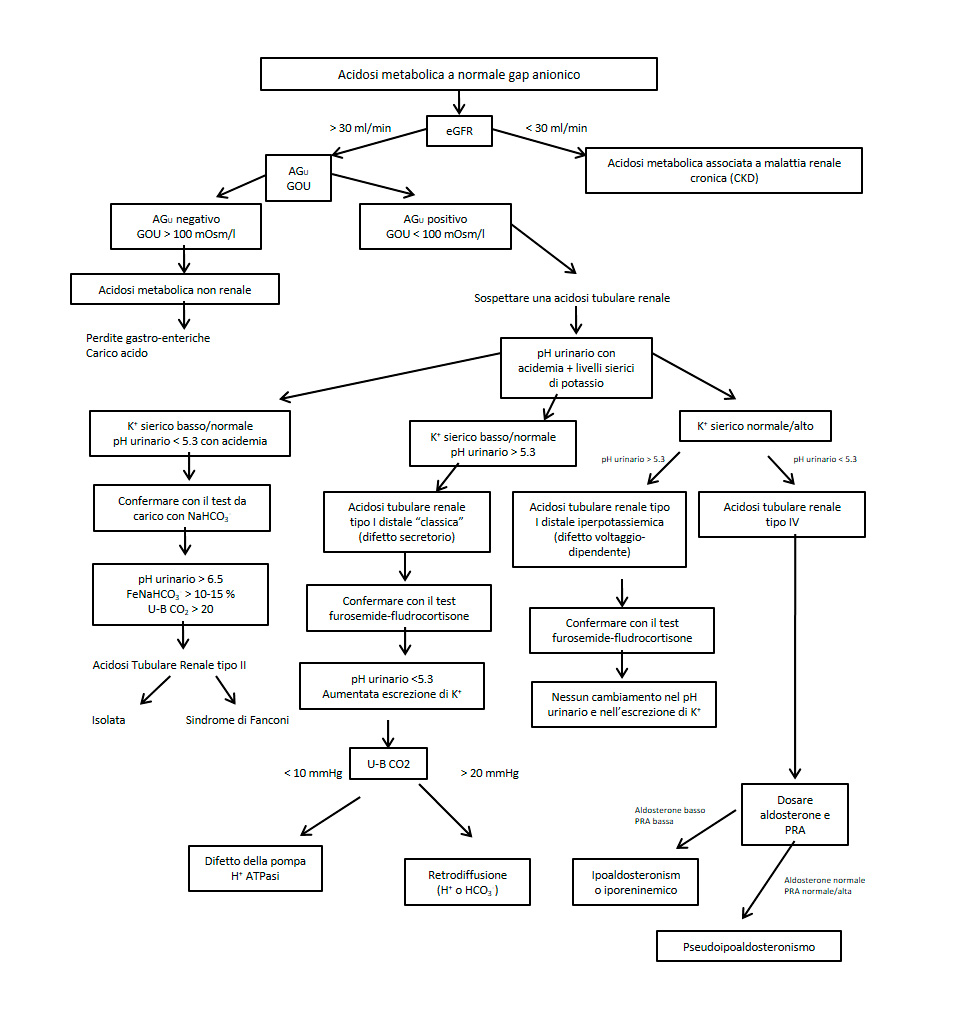{kind=link}
a) ATR prossimale (tipo II)
L’acidosi metabolica nell’ATR prossimale ha due componenti. Una prima componente deriva dall’incapacità del tubulo prossimale di riassorbire il bicarbonato filtrato oltre un livello di soglia di bicarbonati che patologicamente è più basso di quello normale (26 mEq/L); una seconda componente dipende dalla ridotta produzione di NH4+.
Escludendo le forme congenite, le ATR tipo II riconoscono due fasi: una fase iniziale (corrispondente al danno tubulare) con transitorio periodo di perdita urinaria di bicarbonati e conseguente riduzione della bicarbonatemia, ed una seconda fase stazionaria, durante la quale la bicarbonatemia si mantiene stabile su livelli ridotti. Nel tubulo prossimale si verifica un minor riassorbimento di bicarbonato, che giunge al tratto distale superandone le capacità di riassorbimento, con conseguente bicarbonaturia. La fase iniziale è caratterizzata da perdita di sodio associato al bicarbonato, contrazione dell’ECF, iperaldosteronismo secondario, e successivo aumentato riassorbimento di sodio associato prevalentemente a ioni cloro, da cui l’acidosi metabolica ipercloremica. In questa fase, la frazione escreta del bicarbonato è elevata (> 5%) ed il pH urinario è alcalino (> 6), il gap anionico urinario (urinary anion gap, UAG) (Tabella 4) può essere negativo. Il processo continua finché la bicarbonatemia non raggiunge il valore soglia (ridotto) di riassorbimento del tubulo distale (generalmente compreso tra 12-18 mEq/L), raggiunto il quale il bicarbonato filtrato riprenderà ad essere completamente riassorbito. L’ATR tipo II è quindi un disordine autolimitantesi, e l’acidosi sistemica non è progressiva, anche in caso di totale assenza di riassorbimento di bicarbonato prossimale [28] [49]. La seconda componente che concorre a determinare l’acidosi metabolica nell’ATR prossimale, è legata alla ridotta escrezione di NH4+, dovuta all’alcalosi intracellulare nelle cellule tubulari prossimali nei pazienti con forme isolate, ed a disfunzione generalizzata della cellula tubulare prossimale nelle forme inquadrabili nell’ambito della sindrome di Fanconi, nella qualle vi è alterato riassorbimento di fosfati, glucosio, aminoacidi, calcio, citrato, ed acido urico. Le cause più comuni di ATR tipo II sono riassunte in Tabella 2: nell’adulto, tra le più frequenti sono quelle in corso di paraproteinemie e quelle derivanti dall’utilizzo di acetazolamide (inibitore dell’anidrasi carbonica tipo IV), che in condizioni fisiologiche è responsabile della disidratazione dell’H2CO3 a livello luminale [28] [33] [50] (full text). Le alterazioni molecolari in causa nelle forme congenite possono derivare da mutazioni del cotrasportatore sodio-bicarbonato (NBC), con conseguente ridotta escrezione basolaterale di HCO3–, oppure mutazioni dell’anidrasi carbonica tipo II, responsabile a livello intracellulare della formazione di bicarbonato dall’H2O e dalla CO2. Poiché l’enzima è presente anche nelle cellule dell’ultima porzione del tubulo distale, i pazienti con questa mutazione avranno ATR tipo II e tipo I associate (da alcuni autori definita ATR tipo III). L’ATR tipo II può causare osteomalacia, rachitismo e osteopenia nei bambini [47]. La presenza di ATR tipo II deve essere sospettata in ogni paziente con una acidosi metabolica a normale gap anionico e pH urinario <5.3 in presenza di acidemia, in assenza di insufficienza renale cronica (Figura 7).
b) ATR distale (tipo I)
Dipende da un deficit di acidificazione delle urine e può presentarsi in forma iperpotassiemica o ipopotassiemica (Figura 2, Figura 3). La forma ipopotassiemica può essere sostenuta da due meccanismi differenti: ridotta escrezione distale di idrogenioni, su base genetica o acquisita (ad es. Sindrome di Sjögren), oppure aumentata permeabilità delle membrane luminali del tubulo distale, con conseguente retrodiffusione degli idrogenioni. L’ipopotassiemia si instaura in quanto il riassorbimento di sodio, a seguito della diminuita secrezione di H+, avviene prevalentemente in scambio con il K+. L’acidosi metabolica cronica inoltre comporta un ridotto riassorbimento prossimale di bicarbonato e di sodio, mentre l’aumentata perdita renale di sodio innesca un quadro di iperaldosteronismo secondario, con conseguente ipopotassiemia. La forma di ATR tipo I iperpotassiemica (forma voltaggio-dipendente), si associa invece ad una alterazione del normale gradiente transepiteliale negativo, a causa di un ridotto carico di sodio che giunge al tubulo distale. Questo si verifica a seguito di un aumentato riassorbimento prossimale (ad es. nella contrazione del volume extracellulare) e/o come effetto rebound di un ridotto riassorbimento di sodio distale, nel caso in cui il canale del sodio sia bloccato da sostanze come amiloride, triamterene, litio, trimethoprim o pentamidina. I pazienti con anemia falciforme e nefrite lupica possono presentare la variante iperpotassiemica di ATR tipo I. L’acidosi metabolica in definitiva consegue alla minor elettronegatività del tubulo distale, che non favorisce la secrezione di idrogenioni da parte delle cellule intercalate. Lo stesso meccanismo può causare iperpotassiemia, in quanto anche la secrezione distale di potassio è ridotta a causa dell’alterato gradiente transepiteliale [51] [52] (full text) [53]. Le cause più comuni di ATR tipo I sono riassunte in Tabella 2 e Tabella 3. In età pediatrica si presenta più frequentemente come forma ereditaria da mutazione del cotrasporto cloro-bicarbonato (gene SLC4A1) e dell’H+-ATPasi apicale (ATP6V1B1 e ATP6V0A4) [53]. I pazienti affetti da Sindrome di Sjögren possono invece presentare una completa assenza delle pompe H+-ATPasi sulle cellule intercalate, oltre che autoanticorpi contro l’enzima AC II [54]. La nefrolitiasi è spesso associata alle forme di ATR tipo I classica, a causa dell’aumento della calciuria e della fosfaturia, e per la riduzione della citraturia in presenza di pH urinario tendenzialmente alcalino [46]. La presenza di ATR tipo I ipopotassiemica deve essere sospettata in ogni paziente con acidosi metabolica a normale AG e un pH urinario >5.3. Per la diagnosi sono utili i test di acidificazione urinaria con cloruro di ammonio, in quanto la principale caratteristica di questo tipo di ATR è la persistenza di valori di pH urinario > 5.3 anche in corso di acidosi metabolica (Tabella 4). La somministrazione di cloruro di ammonio determina frequentemente nausea e vomito ed è pertanto scarsamente tollerata. Una alternativa a questo test è costituito dalla somministrazione simultanea di furosemide 40 mg e fludrocortisone 1 mg che, nel soggetto sano, inducono una acidificazione urinaria nell’arco di 3-4h [55](Tabella 4). I diuretici dell’ansa aumentano il carico distale di sodio, ed il conseguente aumentato riassorbimento di Na+ a livello del dotto collettore aumenta l’elettronegatività del tubulo, comportando una maggiore secrezione di H+ e K+. Il fludrocortisone invece, agisce aumentando la secrezione di protoni da parte delle cellule intercalate di tipo α. In corso di acidosi metabolica il pH urinario atteso è < 5.3, e sarà ulteriormente ridotto in caso di utilizzo di furosemide, mentre nei pazienti con difetti della pompa H+-ATPasi il pH urinario rimarrà alcalino [55]. Nelle forme di ATR tipo I iperpotassiemiche (difetto voltaggio-dipendente) questo test di acidificazione accentua il disturbo, determinando un incremento dell’escrezione di H + e K+, con un pH urinario che si manterrà alcalino (> 5.3) anche in presenza di un aggravamento dell’acidosi metabolica (bicarbonatemia < 15 mq/l). [55] Il principale difetto che caratterizza l’ATR tipo IV è l’iperpotassiemia causata da un quadro di ipoaldosteronismo. L’aldosterone stimola in modo diretto la secrezione di idrogenioni tramite la pompa H+-ATPasi, il riassorbimento di sodio attraverso gli ENaC (canali epiteliali per il sodio) e la secrezione di potassio da parte delle cellule intercalate (canali ROMK). Inoltre incrementa indirettamente la secrezione di H+ generando una maggiore elettronegatività nel tubulo a seguito del riassorbimento di Na+ [33]. Le principali cause di ATR tipo IV sono riassunte in Tabella 2 Tabella 3. L’acidosi metabolica in tale forma di ATR è solitamente lieve, e la correzione della potassiemia porta spesso alla risoluzione dell’acidosi. L’acidosi metabolica associata alla riduzione della funzione renale è inizialmente ipercloremica, ma con il progredire dell’insufficienza renale può divenire ad elevato gap anionico, a seguito dell’accumulo di acidi organici derivati per lo più dal catabolismo proteico [33] [56] (full text)[57] (full text). Il principale difetto è l’inabilità di produrre e secernere ioni ammonio, con conseguente ridotta escrezione di acidi.
Acidosi metaboliche con gap anionico elevato (accumulo di acidi fissi endogeni o esogeni)
Caratteristica generale di questo gruppo di acidosi metaboliche è l’accumulo di acidi fissi, sotto forma di H+ e del rispettivo anione di accompagnamento (ad es. lattato, beta-OH-butirrato, etc.) (Figura 6). L’accumulo può derivare da alterazioni del normale metabolismo ossidativo, con conseguente aumento della produzione/riduzione consumo di acidi organici (ad es. acidosi lattica, chetoacidosi), dall’introduzione di sostanze con caratteristiche di acidi fissi organici od inorganici (ad es. acido acetilsalicilico), o dalla presenza di sostanze che, pur non essendo di per sé acidi (ad es. metanolo, etanolo, glicole etilenico etc.), vengono successivamente metabolizzate nell’organismo ad acidi organici attraverso vie metaboliche pre-esistenti [27] (full text). Le acidosi metaboliche appartenenti a questo sottogruppo configurano spesso dei quadri clinici rapidamente evolutivi con caratteristiche di emergenza/urgenza. In tali condizioni cliniche il tempestivo riconoscimento dello specifico acido che si sta accumulando, oltre che del processo fisiopatologico sottostante, è fondamentale al fine dell’impostare una terapia corretta in tempi brevi, garantendo così la miglior prognosi del paziente.
1. Chetoacidosi
Scopo della chetogenesi è rendere disponibile per il sistema nervoso centrale substrati energetici idrofili a partire dagli acidi grassi (i chetoacidi) in grado di sostituire il glucosio, quando questo sia carente. La barriera ematoencefalica limita infatti l’accesso degli acidi grassi liberi al parenchima cerebrale. Nel caso dei chetoacidi è invece disponibile di un sistema di trasporto cellulare che consente loro di raggiungere rapidamente il tessuto cerebrale [28]. Il processo di produzione epatica dei chetoacidi prevede due passaggi:
- la formazione di acetil-CoA
- la conversione dell’Acetil-CoA a chetoacidi.
Il più importante substrato per la produzione di acetil-CoA è rappresentato dagli acidi grassi contenuti nei depositi lipidici corporei. La mobilizzazione degli acidi grassi avviene per stimolo da parte degli ormoni contro-insulari in condizioni di relativa carenza insulinica, ovvero durante il digiuno prolungato o scompenso glicemico nel diabete mellito. Substrati alternativi in grado di aumentare la produzione di acetil-CoA sono l’etanolo, in caso di intossicazione acuta, e l’acido acetico derivato dalla fermentazione batterica intestinale di carboidrati non assorbiti (fruttosio e fibre). L’ossidazione degli acidi grassi ad acetil-coA richiede la presenza, come cofattori, di due accettori di protoni costituiti dal nicotinamide adenin-dinucleotide (NAD+) e del flavin-adenin-dinucleotide (FAD+), che vengono ridotti rispettivamente a NADH e FADH2. Poiché questi cofattori sono presenti nei mitocondri in concentrazione minima nella forma ossidata, affinché la chetogenesi proceda è necessario che essi vengano costantemente rigenerati. La principale via di rigenerazione del NAD+ e del FAD+ è costituita dalla fosforilazione ossidativa, che converte l’ADP in ATP. Risulta chiaro quindi, che il consumo di ATP dovuto al “lavoro biologico” che rende nuovamente disponibile ADP, costituisce il principale fattore limitante la fosforilazione ossidativa. Sia durante il digiuno prolungato, che durante la chetoacidosi diabetica, il flusso epatico tende a ridursi, riducendo così anche il “lavoro” epatico, il consumo di ATP e conseguentemente la disponibilità di ADP [28] [58]. In tali condizioni la chetogenesi per procedere richiederà l’attivazione di vie metaboliche in grado di aggirare la fosforilazione ossidativa al fine di rigenerare il NADH ed il FADH2. Questo può avvenire disaccoppiando la respirazione cellulare dalla produzione di ATP. In caso di elevato grado di disaccoppiamento, come si verifica nel corso del digiuno prolungato, la chetogenesi procederà molto rapidamente (fino 1500 mmol/die) e dipenderà direttamente dalla disponibilità di O2 negli epatociti [28]. Vie alternative, che costituiscono analoghi metabolici del disaccoppiamento della fosforilazione ossidativa comprendono la conversione dell’acetoacetato a β-idrossibutirrato, ed il “ciclo futile” del fruttosio 5-fosfato. Tali vie metaboliche, a differenza del disaccoppiamento, non richiedono consumo di ossigeno. Un elevato rapporto NADH/NAD+ può indurre inoltre la lattato-deidrogenasi, determinando accumulo di acido lattico, che può contribuire al carico acido in corso di chetoacidosi. Questo meccanismo può spiegare l’iperlattacidemia che si osserva in alcuni di questi pazienti, anche in assenza di apparente ipotensione o ipoperfusione periferica [28]. Pertanto, nella chetoacidosi diabetica, finché non viene raggiunto un alto grado di disaccoppiamento, un accumulo significativo di chetoacidi è dovuto prevalentemente ad una ridotta velocità di rimozione, piuttosto che ad un’incrementata velocità di sintesi. Due sono i maggiori siti di ossidazione dei chetoacidi, il sistema nervoso centrale (SNC) ed il rene. Il SNC è il principale sito di ossidazione dei chetoacidi, raggiungendo 800 mmol/die in caso di prolungato digiuno [58]. Una riduzione del consumo energetico cerebrale dovuto all’insorgenza di coma, o all’effetto di sedativi tra cui l’etanolo, potrà di conseguenza facilitare l’accumulo di chetoacidi [28][58]. I reni possono rimuovere circa 400 mmol/die di chetoacidi, 250 mEq dei quali vengono ossidati e 150 mEq escreti in associazione a NH4+, garantendo il mantenimento dell’omeostasi acido-base. Se però i chetoacidi vengono eliminati nelle urine in associazione ad un catione diverso dall’ammonio, come avviene ad esempio nella fase poliurica dello scompenso glicemico, o durante la terapia di riespansione volemica, si determinerà una perdita di HCO3– “virtuale”, realizzandosi quindi un quadro di acidosi metabolica a normale gap anionico. Nel corso della chetoacidosi diabetica, tuttavia, il GFR si riduce secondariamente alle perdite di sodio dovute alla diuresi osmotica glucosio-indotta; conseguentemente i chetoacidi tendono ad accumularsi a causa della ridotta ossidazione ed escrezione, determinando un relativo incremento del gap anionico [59]. In minor misura anche il tratto gastro-enterico contribuisce all’ossidazione dei chetoacidi (circa 200-300 mmol/die). In caso di digiuno prolungato o di chetoacidosi diabetica, il lavoro intestinale, e conseguentemente l’ossidazione dei chetoacidi, si riduce considerevolmente a causa del ridotto afflusso ematico, potendo contribuire ulteriormente al loro accumulo [28] [33]. Infine una certa quota viene eliminata con l’espirazione. Infatti l’acido acetoacetico, quando si accumula in elevate concentrazioni, in presenza di bassi livelli di NADH, viene convertito in acetone (volatile) + CO2 [28]. La chetoacidosi diabetica si sviluppa in condizioni di ridotta attività insulinica ed in presenza di elevati livelli degli enzimi contro-insulari (α-adrenergici, glucagone, glucocorticoidi, GH ed ormoni tiroidei). Può essere la manifestazione d’esordio del diabete mellito nel bambino, ma più frequentemente complica un diabete mellito già noto in presenza di fattori precipitanti come infezioni, infarto del miocardio, traumi, pancreatite, abuso di alcool, e terapia insulinica inadeguata. Si manifesta principalmente con poliuria da diuresi osmotica glucosio-indotta, polidipsia per contrazione del volume ematico efficace, astenia, malessere, dolori addominali, perdita di peso (catabolismo della massa magra), progressiva alterazione dello stato di coscienza fino al coma. Il quadro laboratoristico è caratterizzato da iperglicemia, glicosuria, acidosi metabolica ad elevato gap anionico (nella fase di mantenimento), ed aumentata concentrazione sierica di acetoacetato e di β-idrossibutirrato. Il β-idrossibutirrato e l’acetoacetato sono in equilibrio tra loro ma, come già accennato, in caso di accumulo di NADH nel mitocondrio (ad esempio durante il metabolismo dell’etanolo) l’equilibrio della reazione verrà ad essere spostata verso la produzione di β-idrossibutirrato, mentre le concentrazioni di acetoacetato si ridurranno notevolmente. Poiché i test rapidi per la determinazione di chetoni rilevano solo l’acetoacetato e l’acetone, essi possono sottostimare l’effettivo accumulo di chetoacidi. Spesso sono presenti anche deplezione sodica (stimato attorno a 5-10 mmol/Kg), iponatremia ed alterazioni della potassiemia. L’iponatremia dipende da quattro fattori combinati:
- Deficit assoluto di sodio dovuto alla diuresi osmotica (NaU di solito 40-50mEq/L)
- Aumento dell’acqua libera dovuta all’ingestione di liquidi ipotonici ed a liberazione di ADH da stimolo ipovolemico
- Shift extracellulare di H2O, dovuto all’iperosmolarità plasmatica indotta dall’iperglicemia.
- Eventuale presenza di ipertrigliceridemia, che può causare pseudoiponatriemia
La combinazione di tali fattori rende difficilmente prevedibile l’effettivo deficit di sodio rispetto all’effetto legato alla traslocazione di H2O, per cui i “fattori di correzione” normalmente utilizzati per correggere la natremia in corso di iperglicemia possono non essere attendibili, e non è consigliabile la loro utilizzazione [28] [60]. I valori di potassiemia possono essere notevolmente variabili, in base alla causa scatenante o fattori associati (vomito, diarrea, ecc). Generalmente la potassiemia presenta valori leggermente superiori alla norma o nella norma, a causa della relativa carenza insulinica in presenza tuttavia di un deficit assoluto del K+ totale corporeo dovuto alle perdite renali (iperaldosteronismo secondario all’ipovolemia). Dopo la somministrazione di insulina la potassiemia tende a ridursi rapidamente per effetto dello spostamento del potassio all’interno delle cellule. A questo si aggiunge probabilmente un effetto aldosterone-simile dell’insulina a livello del dotto collettore, che può mantenere una potassiuria elevata anche in presenza di deplezione potassica [28]. Il deficit di HCO3– è variabile e può essere sottostimato per la contrazione del volume extracellulare. Il ΔAG/ΔHCO3– può essere pari a 1 nella fase oligoanurica, ma questo rapporto può sottostimare l’effettivo accumulo di acidi per due motivi:
- Una parte dell’idrossibutirrato potrebbe essere stato escreto nelle urine in associazione a Na+o K+ nella precedente fase poliurica.
- Per effetto della contrazione del volume extracellulare, che da un lato aumenta AG (come effetto dell’aumento della concentrazione di albumina), e dall’altro aumenta la concentrazione apparente di HCO3–.
2. Acidosi lattica
Esistono due forme di acido lattico: tipo L e tipo D, a seconda dell’isomero del lattato predominante (levo o destrogiro). L’accumulo dell’isomero L dipende dalla combinazione, in diversa misura a seconda del quadro sottostante, di aumentata produzione ed alterata rimozione dell’acido lattico. L’acido lattico deriva dal metabolismo dell’acido piruvico con una reazione catalizzata dall’enzima lattico-deidrogenasi (LDH), che richiede la conversione di NADH in NAD+ (Figura 8). In condizioni normali, ogni giorno sono prodotti circa 15-20 mmol/kg di acido lattico, principalmente dalla glicolisi o dalla deaminazione dell’alanina [33] [61] (full text). L’acido lattico prodotto viene tamponato dall’HCO3– intracellulare con la formazione di lattato, che a sua volta è riconvertito a piruvato a livello epatico e in minor misura nella corticale renale. L’80% circa del piruvato è ossidato a CO2 + H2O dalla piruvico-deidrogenasi, mentre il 20%, tramite la piruvico-carbossilasi, è convertito a glucosio (Figura 8). Entrambi i processi portano alla rigenerazione dell’HCO3– consumato nel tamponamento iniziale. Queste reazioni richiedono un adeguato metabolismo ossidativo. La concentrazione di acido lattico plasmatico nei soggetti normali è 0.5 – 1.0 meq/L. Un’accelerata produzione di acido lattico si verifica quando la cellula si trova in uno stato di deficit energetico a causa di un alterato funzionamento della catena respiratoria, in presenza o meno di ipossia tissutale (Tabella 5). Si distinguono quindi due forme, rispettivamente tipo A (anaerobica) e tipo B (aerobica) di acidosi L-lattica (Tabella 6). Il blocco della catena respiratoria fa sì che la concentrazione intracellulare di ADP aumenti, mentre il piruvato che si accumula verrà rapidamente convertito in acido lattico. Questa reazione rigenera NAD+, che spinge ulteriormente la glicolisi verso la produzione di piruvato e quindi lattato. Il fattore limitante del processo è costituito dalla progressiva riduzione del pH intracellulare, che blocca l’attività della fosfofruttochinasi. Se il blocco della glicolisi da una parte limita l’ulteriore caduta del pH intracellulare, dall’altro non permette un’ulteriore rigenerazione di ATP, determinando uno stato di grave carenza energetica potenzialmente letale per la cellula, soprattutto negli organi “nobili” come cuore e cervello, che presentano maggiori richieste metaboliche in condizioni basali. La causa più comune di acidosi lattica è l’ipoperfusione sistemica (stati di shock, scompenso cardiaco avanzato, sepsi grave e shock settico, etc), in cui l’ipossiemia costituisce il principale fattore limitante la fosforilazione ossidativa [28] [62](Tabella 5, Tabella 6). Allo stesso modo, un incremento delle richieste energetiche muscolari, tale da eccedere la velocità di rigenerazione di ATP del metabolismo ossidativo aerobico, come quello che si verifica durante le crisi convulsive o a seguito dell’esercizio estremo, può determinare un temporaneo accumulo di acido lattico. Un accumulo di acido lattico può verificarsi anche in presenza di sufficiente apporto di ossigeno, quando sussistano dei deficit del metabolismo ossidativo. La tiamina (vit B1) costituisce il cofattore della piruvico-deidrogenasi (PDH). Uno stato carenziale, generalmente dovuto a prolungato stato di malnutrizione, inattiva l’enzima determinando un accumulo di piruvato, che viene convertito a lattato. Tale deficit è particolarmente pericoloso in corso di risoluzione della chetoacidosi da digiuno (sospensione alcool, rialimentazione dopo digiuno prolungato) in quanto può precipitare il tessuto cerebrale, che fino a quel momento utilizzava come combustibile preferenziale i corpi chetonici, in uno stato di improvvisa carenza energetica (sindrome di Wernicke-Korsakoff). L’acidosi lattica in assenza di ipossia può determinarsi anche in presenza di fattori che interagiscano direttamente con la catena protonica (es. carenza di riboflavina, cianidi ed antidepressivi triciclici), o a causa di un eccessivo grado di disaccoppiamento della catena respiratoria.
{kind=link}
Il disaccoppiamento consiste nel trasporto di H+ attraverso la membrana interna dei mitocondri senza produzione di ATP. Questo processo può avvenire attraverso due meccanismi:
- Effetto di proteine disaccoppianti (UCP), che sono canali protonici espressi sulla membrana mitocondriale non associati all’ADP-fosforilasi
- Effetto di acidi deboli in grado di attraversare la membrana mitocondriale e che agiscono come trasportatori di protoni (fenformina, metformina e salicilati)
Tale processo è presente fisiologicamente in quanto garantisce alcuni vantaggi quali:
- nel fegato, in presenza di bassi livelli di insulina, garantisce un rifornimento di NAD+ rivolto alla produzione di corpi chetonici che possono essere ossidati nel cervello e nel rene durante il digiuno prolungato.
- permette una più rapida metabolizzazione dell’etanolo
- avrebbe un ruolo protettivo dal danno da radicali liberi dell’ossigeno
- regola la termogenesi (almeno nell’animale), controllando il peso corporeo e l’accumulo dei trigliceridi nel tessuto adiposo.
Tuttavia un eccessivo disaccoppiamento può determinare una carenza di ATP, con conseguente produzione di acido lattico e danno cellulare [28]. Numerosi farmaci, in primo luogo le biguanidi, possono interagire con la catena respiratoria determinando acidosi lattica [62]. I meccanismi possono variare da molecola a molecola, ma generalmente tali sostanze agiscono come trasportatori di protoni attraverso la membrana mitocondriale, dissipando il gradiente protonico a livello della membrana mitocondriale interna. La metformina normalmente non determina acidosi lattica, a meno che non si accumuli in presenza di un ridotto GFR. In tal caso la dialisi può rimuoverla efficacemente, in considerazione del basso peso molecolare e del ridotto legame farmaco-proteico, ma a causa dell’alto volume di distribuzione e della cinetica bi-compartimentale sono consigliati trattamenti prolungati [62] [63] (full text) che tengano conto del possibile rebound dei livelli plasmatici del farmaco. Il principale organo i cui viene metabolizzato il lattato è il fegato. Una significativa riduzione della massa di tessuto funzionante in caso di cirrosi avanzata o esteso sovvertimento neoplastico, può costituire un’ulteriore causa di accumulo di lattati, soprattutto se associata anche ad un incremento della loro produzione [28]. L’acidosi D-lattica è una sindrome relativamente rara, che può verificarsi in pazienti con malassorbimento intestinale di carboidrati (sindrome dell’intestino corto, bypass digiuno-ileali) e dismicrobismo intestinale con presenza di flora batterica fermentante che produca elevati quantitativi di D-acido lattico. Diversi fattori potrebbero contribuire alla patogenesi di tale forma di acidosi metabolica, tra cui il basso pH intestinale, l’uso prolungato e/o eccessivo di antibiotici e probiotici, ed infine la ridotta escrezione renale. I segni ed i sintomi neurologici, rapidamente normalizzati dal trattamento dialitico, comprendono alterazioni dello stato mentale, disartria, atassia, incoordinazione motoria e si associano ad acidosi metabolica a gap anionico aumentato, con livelli di L-lattato nella norma, in quanto l’isomero destrogiro del lattato non viene misurato nelle comuni determinazioni di laboratorio [28] [33] [64] (full text).
3. Intossicazione da alcoli e glicoli
Appartengono a questo gruppo le intossicazioni acute da alcoli (ad es. metanolo ed etanolo) e da glicoli o dialcoli (ad es. glicole propilenico e glicole etilenico) [65]. L’intossicazione da alcoli avviene tipicamente per ingestione, ma sono descritti alcuni casi per inalazione (metanolo), assorbimento transdermico (metanolo e glicole propilenico), o per via endovenosa (glicole propilenico). Il problema clinico è dovuto sia all’acidosi che alla possibile tossicità sistemica e renale degli intermedi che derivano dal metabolismo di tali sostanze (ad es. formaldeide e formato nel caso del metanolo, glicolaldeide nel caso del glicole etilenico) [66] (full text) (Figura 9 a,b). Il sospetto deve sempre essere posto in presenza di un’acidosi metabolica ad elevato AG, quando non sia evidente dall’anamnesi e dal contesto clinico l’acido accumulato all’origine del disturbo. Risulta fortemente orientativo per l’ingestione di alcoli un elevato OG. L’incremento dell’osmolalità plasmatica tuttavia tende a normalizzarsi man mano che l’alcol o il glicole venga metabolizzato ad acido organico, e presenta pertanto un andamento temporale inverso rispetto all’aggravamento dell’acidosi metabolica. Il riscontro di aumentato OG nelle intossicazioni da alcoli/glicoli dipende quindi dalla cinetica dei singoli composti e dal momento di presentazione [10] (full text). La chetoacidosi alcolica è presente in una minoranza delle intossicazioni acute da etanolo (< 10% dei casi), ed è più frequente in caso di ingestione di forti quantità di alcol su di un substrato di alcolismo cronico associato a malnutrizione, deplezione volemica (vomito) ed epatopatia cronica [10] (full text). Dopo l’ingestione l’etanolo raggiunge il circolo portale e viene rapidamente metabolizzato ad acetil-CoA, al fine di evitare gli effetti depressivi sul sistema nervoso centrale. L’enzima chiave della via metabolica è l’alcool deidrogenasi (Figura 9a). Analogamente a quanto avviene nel digiuno prolungato, la chetogenesi è promossa dall’attività degli ormoni controinsulari in presenza di bassi livelli di insulina e ridotte riserve di glicogeno. Tuttavia, nel caso della chetoacidosi alcolica l’accumulo di acetil-CoA, che si forma dal metabolismo dell’etanolo, costituisce il principale substrato per la formazione dei corpi chetonici. Pertanto la chetogenesi nell’intossicazione da etanolo procederà molto più rapidamente rispetto a quella che avviene nel digiuno prolungato. Infatti, mentre deve trascorrere un certo intervallo di tempo prima che gli acidi grassi a catena lunga inizino ad essere metabolizzati a chetoacidi, il metabolismo dell’etanolo è immediato, e la velocità della reazione dipende solo dalla disponibilità dei cofattori ADP e NAD+ [28] [10] (full text). Durante l’ossidazione dell’etanolo a seguito della produzione di chetoacidi si verifica quindi un aumentato del rapporto NADH:NAD+ che determina una induzione della lattico-deidrogenasi (Figura 10) con il relativo aumento della concentrazione di L-lattato a scapito del piruvato, cui segue la ridotta conversione del piruvato a glucosio. Questo è verosimilmente il meccanismo fisiopatologico che rende ragione dell’ipoglicemia nei pazienti con etilismo cronico (malnutrizione e ridotte riserve epatiche di glicogeno) in corso di chetoacidosi alcolica. L’alterato rapporto NADH/NAD+ è anche alla base anche della formazione di β-idrossibutirrato come chetoacido prevalente che, come già accennato, può determinare dei falsi negativi al test rapido per la presenza di corpi chetonici. La chetoacidosi alcolica risulta più severa nei soggetti malnutriti (maggiore rischio di ipoglicemie, maggiore deplezione di Na+/volume), nei quali deve sempre essere considerato anche un possibile deficit associato di tiamina, che può avere effetti gravi nelle fasi di risoluzione della chetoacidosi [28]. La chetoacidosi alcolica si presenta tipicamente con elevato AG e, a differenza della chetoacidosi diabetica, le forme ipercloremiche sono rare anche dopo infusione di soluzione salina. Il riscontro di acidosi metabolica ad elevato AG isolata è comune, ma più frequentemente sono presenti disturbi misti (acidosi metabolica + alcalosi respiratoria, acidosi metabolica + alcalosi metabolica, disturbi misti tripli). L’osmolalità plasmatica è normalmente conservata, in quanto l’acidosi si sviluppa dopo che l’etanolo è stato metabolizzato. Iponatremia, ipokaliemia o iperkaliemia, ed ipofosfatemia possono comunemente associarsi al quadro di acidosi. L’ipoglicemia è un reperto frequente soprattutto nei pazienti con malnutrizione cronica. La terapia consiste nel supporto delle funzioni vitali e della pervietà delle vie aeree in caso di grave compromissione del sensorio, nella somministrazione di liquidi e glucosio, nella correzione di eventuali alterazioni degli elettroliti e nella supplementazione di tiamina. Il metanolo viene utilizzato come additivo della benzina e come solvente, può essere presente nei fluidi antigelo, ed è a volte utilizzato come adulterante del vino per aumentarne la gradazione alcoolica. Il metanolo ha di per sé una tossicità limitata, ma sono particolarmente tossici i suoi metaboliti. Il metanolo viene infatti metabolizzato dall’alcool-deidrogenasi in formaldeide, e successivamente in acido formico dall’aldeide-deidrogenasi, con una reazione che, analogamente all’ossidazione dell’etanolo, riduce il NAD+. L’acidosi ad elevato AG deriva dall’accumulo di formato e di lattato, ma un problema è rappresentato dall’accumulo di formaldeide, che risulta estremamente tossica, anche a livello retinico, per basse concentrazioni. La sintomatologia è costituita da sensazione di ebbrezza, atassia, disartria e successivamente cecità, dolore addominale (si associa spesso a pancreatite acuta), e progressivo peggioramento dello stato di coscienza fino al coma, che si manifesta nell’arco di 6-24 ore dall’ingestione. Una complicanza rara è rappresentata dalla necrosi del putamen, che determina la comparsa di rigidità, tremori, amimia e distonia [10] [10] (full text). La tossicità da metanolo può essere aggravata da concomitante deficit di tetraidrofolato, che può limitare la velocità di conversione dalla formaldeide ad acido formico. Il glicole etilenico è utilizzato prevalentemente nei fluidi antigelo, nel liquido dei freni, e come solvente nelle vernici ed inchiostri industriali. Il metabolismo è sempre dovuto all’azione sequenziale dell’alcool-deidrogenasi e dell’aldeide-deidrogenasi, che formano rispettivamente glicolaldeide e acido glicolico, una parte del quale è successivamente convertito ad acido ossalico. L’acido glicolico è il principale responsabile dello sviluppo di acidosi metabolica, ma può anche interagire con la catena protonica boccando la respirazione cellulare. L’acido ossalico invece precipita nei tessuti (cuore, rene e SNC) come ossalato di calcio, e può determinare disfunzione multiorgano ed ipocalcemia. L’escrezione urinaria dell’ossalato o la sua precipitazione nei tessuti può far sottostimare il contributo dell’acido ossalico allo sviluppo di acidosi. Le manifestazioni inziali sono analoghe all’intossicazione da metanolo o etanolo, mentre nelle fasi tardive può comparire progressivamente ipocalcemia, disfunzione cardio-respiratoria ed infine insufficienza renale entro 12-72 ore dall’assunzione. Oltre al trattamento standard di supporto delle funzioni vitali, la somministrazione di tiamina e piridossina possono facilitare la conversione dell’acido glicolico e del gliossalato a composti meno tossici quali l’α-idrossil-β-ketoadipato. Il glicole dietilenico è un composto industriale presente nel liquido dei freni o come adulterante nella produzione di superalcolici e farmaci. Presenta un metabolismo analogo a quello del glicole etilenico, ma rispetto a quest’ultimo presenta una maggiore tossicità epatica e pancreatica. L’intossicazione è rara, ma presenta elevata mortalità (circa 90%). Il glicole propilenico può essere presente come eccipiente in alcuni farmaci (es. diazepam, lorazepam, fenobarbital, etomidato, fenitoina). Normalmente il glicole propilenico è escreto immodificato nelle urine poiché l’alcol-deidrogenasi presenta una bassissima affinità per esso. Se tuttavia il farmaco a cui è associato viene somministrato in infusione continua in quantità elevate (ad es. lorazepam nel trattamento del delirium tremens), l’aumento delle concentrazioni ematiche può far sì che una parte venga convertita in lattaldeide e successivamente acido lattico. Inoltre poiché il glicole propilenico si trova generalmente nelle due forme isomeriche e l’aldeide deidrogenasi presenta affinità prevalentemente per l’isomero L, si verifica un progressivo accumulo di D-lattaldeide, che è la principale responsabile degli effetti tossici (convulsioni, instabilità emodinamica, insufficienza renale). Contemporaneamente può determinarsi accumulo di acido D-lattico attraverso una riduzione della D-aldeide da parte di una via metabolica alternativa alla lattico-deidrogenasi, che ha come cofattore il glutatione. Oltre al quadro di acidosi metabolica è descritta l’insorgenza di insufficienza renale acuta e disfunzione del tubulo prossimale associate a glicole propilenico, ma la patogenesi non è chiara. Generalmente la sospensione del farmaco è sufficiente a far regredire il quadro. La terapia di queste forme di intossicazione da alcoli/glicoli si basa sull’inibizione dell’alcool-deidrogenasi, al fine di ridurre la formazione dei metaboliti tossici, mediante la somministrazione controllata di etanolo (l’etanolo presenta un’affinità per l’alcol-deidrogenasi 10-20 volte superiore rispetto agli altri alcoli), o di uno specifico inibitore dell’enzima, il fomepizolo (affinità 500-1000 volte superiore rispetto all’etanolo). Quest’ultimo anche se notevolmente più costoso, quando disponibile, è da preferire rispetto all’etanolo, in quanto più maneggevole e gravato da meno effetti collaterali. Quando si utilizza l’etanolo, questo deve essere somministrato in infusione continua, mantenendo circa 100 mg/dL come valore target di etanolemia al fine di saturare completamente l’alcol-deidrogenasi. In alcuni casi è stata raccomandata la somministrazione di alcali al fine di promuovere l’eliminazione del formato e del glicolato [10] (full text) [67] [68] [69] [70]. L’efficacia di tale approccio non è tuttavia dimostrata, e potrebbe essere favorita la precipitazione di ossalato di calcio nei tessuti. La supplementazione con acido folico, piridossina o tiamina può essere utile in casi specifici. L’emodialisi, sia intermittente che continua, è estremamente efficace nel rimuovere alcoli e glicoli, che altrimenti tendono ad essere riassorbiti a livello tubulare. Il tempo di trattamento necessario a ridurre la concentrazione del metanolo (o di un altro alcool) a livelli < 5 mmol/L, può essere stimato dalla formula [71]:
{kind=link}
{kind=link}
T = -V ln(5/A)/0,06k
nella quale V è il volume di acqua totale corporea stimato dalla formula di Watson, A è la concentrazione iniziale in mmol dell’alcol, e k è l’80% della clearance del filtro per l’urea in ml/min. Non ci sono attualmente evidenze sulla superiorità della dialisi rispetto alla terapia medica nel ridurre la mortalità [10] (full text). La Società Americana di Tossicologia raccomanda il ricorso all’emodialisi in presenza di acidosi metabolica ( pH <7,3), disturbi visivi, insufficienza renale e/o alterazioni elettrolitiche che non rispondano alla terapia convenzionale, oppure quando siano documentati valori ematici elevati dell’alcol/glicole responsabile dell’intossicazione (metanolo >50 mg/dl, glicole etilenico >8 mmol/dL, glicole dietilenico >1.34 mg/kg) [68].
4. Intossicazione da Salicilati
L’aspirina viene rapidamente convertita ad acido salicilico nell’organismo. Sebbene non esista una precisa correlazione tra valori ematici di acido salicilico e sintomatologia, la maggior parte dei pazienti mostrano segni di intossicazione già a livelli di 50 mg/dL (intervallo terapeutico 20-35 mg/dL). L’acido salicilico presenta un elevato legame alle proteine plasmatiche (90%) e viene eliminato prevalentemente a livello renale per secrezione tubulare. Una piccola percentuale è glucuronata ed escreta per filtrazione glomerulare [33]. In caso di intossicazione si verifica una saturazione del legame proteico con relativo aumento della quota libera. Allo stesso tempo, la contrazione del GFR, dovuta all’inibizione della ciclossigenasi, riduce l’escrezione del farmaco. I sintomi comprendono acufeni, vertigini, nausea, vomito, diarrea, progressiva alterazione dello stato di coscienza fino al coma ed edema polmonare non cardiogeno, e dipendono principalmente dalla tossicità dell’acido salicilico a livello intracellulare. Infatti analogamente ad altri acidi organici, l’acido salicilico può agire come trasportatore di protoni, determinando disaccoppiamento della fosforilazione ossidativa, carenza di ATP e aumentata produzione di acido lattico [28]. Il disturbo acido-base più frequente e precoce in corso di intossicazione da salicilati è l’alcalosi respiratoria, verosimilmente a seguito di uno stato di ipossiemia relativa che si verifica a livello dei centri respiratori. Nelle forme più gravi è possibile riscontrare un’acidosi metabolica generalmente di lieve-moderata entità ad elevato AG, dovuta all’accumulo di diversi acidi organici quali acido lattico, chetoacidi ed acido salicilico. La tossicità dell’acido salicilico dipende dalla quota che entra nelle cellule. Poiché solo la forma indissociata dell’acido salicilico, essendo apolare, può attraversare le membrane, la tossicità dipende dal grado di dissociazione. Considerando la sua costante di dissociazione (pKa = 3), quanto più il pH si riduce, tanto più il salicilato tenderà ad entrare nelle cellule. Il trattamento deve quindi essere orientato al mantenimento di un pH tendenzialmente alcalino. Nelle forme in cui è presente acidosi metabolica può essere considerata la somministrazione di bicarbonati, facendo attenzione al rischio di ipercorrezione dovuta alla coesistente alcalosi respiratoria. Nelle forme con grave alcalosi respiratoria, può essere considerata la somministrazione estemporanea di dosi ridotte di acetazolamide, in modo da favorire l’escrezione dei salicilati attraverso l’alcalinizzazione delle urine e la conseguente ridotta retrodiffusione del salicilato [28]. La somministrazione di acetazolamide non è esente da rischi in quanto può competere con il salicilato per il legame proteico, aumentandone la quota libera, oltre ad aggravare l’acidosi intracellulare favorendo l’ingresso dell’acido salicilico nelle cellule. La dialisi può essere indicata nelle forme particolarmente gravi (valori ematici >80 mg/dL) con compromissione dello stato di coscienza, nei quali sia presente un’elevata quota libera che può essere rimossa dal trattamento. In caso di gravi intossicazioni (S. di Reye) può essere indicata anche la somministrazione di glucosio al fine di prevenire la neuroglucopenia [28] [33].
5. Acidosi metabolica da altri acidi organici
Il tuolene è presente come solvente in numerose colle e può essere inalato accidentalmente o volontariamente (glue sniffing). Di per sé non è un acido, ma viene rapidamente convertito dal fegato ad acido ippurico. A differenza della maggioranza degli acidi organici l’acido ippurico viene sia filtrato che secreto, ed è pertanto rapidamente eliminato nelle urine in associazione al sodio o al potassio, determinando un’acidosi metabolica a normale AG. La diagnosi può essere confermata dall’anamnesi in presenza di un gap anionico urinario positivo ed un elevato gap osmolale urinario [33]. L’acidosi da acido piroglutammico (5-oxiprolina) normalmente viene associata ad errori congeniti del metabolismo ( deficit glucosio-6-fosfato deidrogenasi), tuttavia si può verificare anche in pazienti critici come effetto di un esteso danno ossidativo, o in caso di intossicazione da acetaminofene. L’acido piroglutammico è un intermedio del metabolismo del glutatione. Normalmente il glutatione viene sintetizzato a partire dall’acido glutammico, dalla cisteina e dalla glicina, attraverso un ciclo, che ha come enzima chiave la γ-glutamil-cistein sintetasi, che a sua volta è inibita dal glutatione con un meccanismo a feed-back negativo. Quando il glutatione si riduce drasticamente a seguito della produzione di ROS (intossicazione da paracetamolo, sepsi), aumenta l’attività della γ-glutamil-cistein sintetasi e può accumularsi acido piroglutammico. Questo è ancora più probabile in presenza di farmaci che inibiscano la 5-oxiprolinanasi (vigabatrin, flucloxacillina), che normalmente riconverte l’acido piroglutammico in glutammato [28].
Principi di terapia delle acidosi metaboliche
I principi generali del trattamento dell’acidosi metabolica sono illustrati in Tabella 7. L’acidemia acuta grave (pH <7.1) può teoricamente determinare una serie di effetti negativi (Tabella 8), tra i quali depressione della contrattilità miocardica, aumentata suscettibilità alle aritmie, vasoplegia e ridotta risposta ai vasopressori, insufficienza respiratoria, vasocostrizione del circolo polmonare, ridotta ossigenazione dei tessuti (effetto Bohr), edema cerebrale ed insulino-resistenza [1] (full text) [5] (full text) [72]. La maggioranza dei clinici tende a somministrare bicarbonati nel caso in cui i valori di pH siano <7.20, indipendentemente dall’eziologia del disturbo [73], al fine di prevenire complicanze direttamente connesse all’acidosi, ed in particolare per garantire una maggiore stabilità emodinamica del paziente. Tale approccio è basato su studi fisiopatologici effettuati in vitro ed in modelli animali, anche se non è chiaro se la correzione dell’acidemia attraverso la somministrazione di basi sia effettivamente in grado di modificare la prognosi nell’uomo. La somministrazione di soluzioni alcalinizzanti deve essere attentamente valutata sulla base del processo fisiopatologico alla base dell’acidosi, e valutando in parallelo la possibile insorgenza di complicanze correlate alla terapia stessa [74] (full text)[75] (full text). Nell’acidosi lattica non esistono attualmente evidenze robuste sulle indicazioni alla somministrazione di bicarbonati. Sebbene teoricamente la correzione di una acidosi grave (pH <7.10) possa migliorare l’emodinamica del paziente, non esistono studi clinici che ne dimostrino l’effettiva efficacia in termini di riduzione della mortalità [62] [74] (full text) [75] (full text). Alcune evidenze cliniche e sperimentali sottolineano i possibili effetti collaterali secondari alla somministrazione di bicarbonato, quali peggioramento dell’acidosi intracellulare per aumento della pCO2, ipopotassiemia e ipocalcemia (calcio ionizzato), che a loro volta possono deprimere l’attività dei muscoli respiratori e la funzione miocardica, oltre a favorire le aritmie (Tabella 9) [62][75] (full text). Il bicarbonato può inoltre riattivare la fosfofruttochinasi (inibita dall’acidosi), aumentando ulteriormente la velocità di produzione del lattato, oltre a promuovere la formazione di radicali liberi, citochine proinfiammatorie ed attivare l’apoptosi. Ulteriori rischi della terapia alcalinizzante possono essere rappresentati dal sovraccarico di volume, da ipernatremia e iperosmolarità, aumento della PaCO2 ed ipercorrezione dell’acidosi, con conseguente alcalosi metabolica [1] (full text) [74] (full text) [75] (full text). Al fine di ridurre il rischio di aumento della produzione di CO2 a seguito della somministrazione di bicarbonati sono stati messe a punto altre soluzioni alcalinizzanti che non presentano tale inconveniente, quali il CarbicarB (soluzione equimolare di sodio bicarbonato e sodio carbonato) ed il THAM (tris-idrossimetilamino-metano). Queste soluzioni sono state sperimentate prevalentemente nell’acidosi lattica, ma hanno avuto una ridotta diffusione per la presenza di effetti collaterali, e per mancanza di studi controllati che ne documentassero un migliore profilo di sicurezza rispetto al bicarbonato, a fronte di un costo maggiore [62]. Sebbene l’iperlattacidemia sia riconosciuta, secondo diversi autori, come un importante fattore prognostico negativo (mortalità del 90% nei pazienti nei quali persista acidosi lattica grave a 24 ore dall’inizio della terapia) [76] [77] [78] (full text) [79] (full text), non è attualmente definito quanto il trattamento dell’acidemia in sé, possa influenzare direttamente la prognosi [62]. In realtà l’acidosi lattica è più spesso in realtà l’epifenomeno di un grave deficit energetico cellulare, dovuto ad un ampio spettro di cause sottostanti. Le misure più efficaci nella gestione dell’acidosi lattica devono quindi essere rivolte in primo luogo al problema di base, ed in particolare all’ottimizzazione della perfusione e dell’ossigenazione tissutale, e sia l’algoritmo dell’ACLS (Advanced Cardiac Life Support), che le linee guida Surviving Sepsis Campaign non raccomandano al momento l’utilizzo precoce di alcali [75] (full text). Nei pazienti con acidosi lattica l’impiego di elevate quantità di cristalloidi al fine di trattare la patologia sottostante (shock emorragico, trauma, sepsi, ecc) può associarsi all’insorgenza di acidosi ipercloremica ed ulteriore peggioramento della prognosi (vedi più avanti). L’obbiettivo del trattamento della chetoacidosi diabetica si è storicamente incentrato sulla rapida risoluzione del deficit insulinico, oltre che sulla correzione dell’ipovolemia [80]. Nonostante esistano numerose linee guida sul trattamento della chetoacidosi, la maggior parte delle raccomandazioni si basano su casistiche quantitativamente limitate e pochi studi randomizzati con ampia variabilità nella tipologia dei pazienti, nei valori soglia e nelle modalità di somministrazione del bicarbonato, insufficienti a fornire significativi gradi di evidenza [81] (full text). L’indicazione alla somministrazione di bicarbonati nella chetoacidosi diabetica si è progressivamente ristretta nel corso degli anni, sulla base di studi che non ne hanno confermato l’efficacia, a fronte talvolta di una maggior frequenza di eventi avversi e tempi di degenza più lunghi [80] [81] (full text). I pazienti pediatrici, in particolare, sarebbero a maggior rischio (almeno 4 volte) di sviluppare edema cerebrale in corso di terapia con bicarbonato [82] (full text). Sulla base di tali studi attualmente la maggioranza degli esperti concorda sulla non indicazione dell’infusione di alcali nei pazienti in età pediatrica e per valori di pH al di sopra di 6.9 [28] [58] [80] [81] (full text). Nella chetoacidosi diabetica la somministrazione di bicarbonati presenta due benefici teorici: la stabilizzazione emodinamica in pazienti con acidemia grave, e la prevenzione dell’insorgenza di un’acidosi ipercloremica, che si osserva frequentemente (50% dei casi a 2 ore dall’inizio della terapia e fino al 90% dei casi a 8-20 ore) [83] [84] nelle fasi di recupero dello squilibrio metabolico. Sebbene alcuni studi caso-controllo abbiano dimostrato una più rapida risoluzione dell’acidemia dopo somministrazione di bicarbonati, l’effetto è risultato transitorio, e non sono state dimostrate differenze di outcome [80]. Dal punto di vista della stabilità emodinamica, nei tre studi disponibili, il bicarbonato non è risultato vantaggioso rispetto alla soluzione salina [85] [86] [87]. È tuttavia necessario sottolineare il fatto che mancano in letteratura studi nel soggetto adulto che abbiano valutato i parametri emodinamici e la risposta alla terapia alcalinizzante in pazienti critici con severa compromissione emodinamica e pH <6.85, condizioni cioè nelle quali verosimilmente gli effetti emodinamici dell’acidemia potrebbero essere più evidenti. In una popolazione pediatrica con valori di pH <6.73 il quadro di acidemia si è risolto indipendentemente dalla somministrazione di bicarbonati [81] (full text) [88]. Questi studi aprono quindi una prospettiva ulteriormente critica rispetto all’indicazione dei bicarbonati nella chetoacidosi diabetica. Poiché l’utilità della somministrazione di bicarbonati nel trattamento della chetoacidosi diabetica rimane controversa, nell’adulto l’indicazione posta unicamente in base al valore del pH arterioso è probabilmente insufficiente a garantire un adeguato margine di sicurezza per una terapia potenzialmente dannosa. In questo senso si raccomanda un approccio integrato da una valutazione più ampia dello stato acido-base, che permetta di individuare i pazienti a rischio di sviluppare rapido peggioramento dell’acidemia a seguito di un’ulteriore riduzione della bicarbonatemia. La somministrazione di bicarbonati può quindi essere presa in considerazione in pazienti con acidemia grave in cui i sistemi di compenso siano in fase di esaurimento (massimo stato di iperventilazione e rischio di fatica/esaurimento dei muscoli respiratori), o in cui sia attesa una lenta ossidazione dei corpi chetonici a causa di una grave compromissione dello stato di coscienza o insufficienza renale di grado avanzato precedente all’insorgenza della chetoacidosi [58]. L’acidosi a normale AG, o ipercloremica, è di frequente riscontro nei pazienti critici (circa il 30% delle acidosi metaboliche) a seguito della perdita di bicarbonati dal tratto gastroenterico, o per la somministrazione di forti quantità di soluzioni contenenti cloro per il trattamento dell’ipovolemia e dello shock [8] (full text). Il sovraccarico di cloro, che può verificarsi nei pazienti in terapia intensiva dipende sia da una ridotta escrezione che da un aumentato apporto dovuto all’uso di soluzioni a concentrazione di cloro sovrafisiologiche. In questo senso è stato ipotizzato che l’aumentato apporto di cloro agisca a livello della macula densa determinando una riduzione del GFR attraverso il feedback tubulo-glomerulare. Alcuni studi in modelli animali e nell’uomo hanno dimostrato una riduzione del flusso plasmatico renale dopo somministrazione di soluzioni a contenuto non fisiologico di cloro [7] (full text). La somministrazione di basi è spesso indicata in questa tipologia di acidosi, ma non esistono studi che diano precise indicazioni sui valori soglia o le modalità di somministrazione. Allo stesso modo non è chiaramente stimato il rischio di complicanze connesse alla terapia. Il valore prognostico dell’acidosi ipercloremica da somministrazione di fluidi e gli eventuali risvolti terapeutici di essa sono attualmente oggetto di dibattito. Sebbene in modelli sperimentali l’acidosi da “diluizione” si associ ad un incremento della mortalità, l’impatto prognostico di essa nell’uomo non è stato del tutto chiarito. Una recente meta-analisi [89] (full text) ha dimostrato un rischio significativamente aumentato di sviluppare insufficienza renale acuta, aumentato fabbisogno trasfusionale e prolungamento del tempo di ventilazione meccanica, ma non un aumento della mortalità, nei pazienti trattati con soluzioni ad “alto” contenuto di cloro, come ad esempio la salina allo 0.9%. Nonostante alcune limitazioni dello studio, quali il ridotto numero di pazienti inclusi nei vari studi considerati e l’eterogeneità delle popolazioni in esame, gli autori concludono raccomandando l’impiego di soluzioni a “bassa” concentrazione di cloro (<111mmol/l). Non vengono però forniti dati circa il trattamento di queste forme di acidosi una volta instauratesi. Non è chiaro infatti se l’insorgenza di complicanze (in primis l’insufficienza renale) ed il peggioramento della prognosi dipenda direttamente dall’acidosi e dal sovraccarico di cloro, o piuttosto dal concomitante sovraccarico di fluidi [8] (full text). Nelle forme croniche di acidosi metabolica la terapia è mirata alla prevenzione degli effetti a lungo termine dello squilibrio. Nelle acidosi renali tubulari prossimali e distali la maggioranza degli esperti concorda sull’indicazione alla somministrazione di basi. La somministrazione di bicarbonati è risultata inoltre efficace nel garantire un normale profilo di accrescimento staturale nei bambini con acidosi tubulare [90]. Mancano tuttavia in letteratura studi sulle modalità ed i targets terapeutici. Va tenuto presente che anche in questo caso un’ipercorrezione può determinare bicarbonaturia, sia nelle forme prossimali che in quelle distali, che può facilitare la precipitazione tubulare di fosfato di calcio. La somministrazione di citrato al posto del bicarbonato, soprattutto nelle forme distali, che si associano ad ipocitraturia, può ridurre questo rischio [28]. Nell’insufficienza renale cronica, le numerose evidenze relative all’aumentata incidenza di complicanze e della progressione dell’insufficienza renale connessi all’acidosi metabolica supportano la somministrazione di bicarbonati [91] [92] (full text). Il rischio teorico di peggioramento del controllo pressorio e del sovraccarico di volume secondario alla terapia con bicarbonato di sodio non sembra confermato dagli studi effettuati ed appare generalmente ben tollerato in pazienti con diuresi conservata. Un’altra possibile complicanza della terapia alcalinizzante in corso di insufficienza renale cronica è costituita dall’aumentato rischio di calcificazioni vascolari. Quest’ipotesi è sostenuta da studi in vitro e su modelli animali che ipotizzavano un ruolo protettivo dell’acidosi metabolica rispetto alle calcificazioni extravascolari. Mancano tuttavia in questo senso studi nell’uomo che supportino tale ipotesi [93] (full text) [94] (full text). La somministrazione di bicarbonati è indicata per valori < 22 mEq/L. Normalmente la dose iniziale è di 10-20 mEq/die, incrementando successivamente la dose sulla base dei valori di bicarbonatemia (per un aumento di 0,1 mEq/Kg/die è atteso un aumento di 0,33 mEq della bicarbonatemia) [91] [92] (full text). Una valida alternativa nei pazienti intolleranti al bicarbonato è costituita dal sodio citrato. Anche i chelanti del fosforo, come il calcio acetato o carbonato o il sevelamer carbonato, possono contribuire all’apporto di basi. Anche se attualmente non esiste ancora un consenso assoluto rispetto ai valori target di bicarbonatemia, viene comunemente raccomandato di mantenere livelli di 24-26 mEq/L. Quando è indicata la somministrazione di bicarbonati per via parenterale, è necessario tenere presenti alcuni concetti sulla cinetica dei bicarbonati. Nella comune pratica clinica si tende a considerare come spazio di distribuzione del bicarbonato il volume dell’acqua totale corporea. Il deficit di bicarbonati può quindi essere calcolato con la formula:
∆HCO3– = 0,5*(Bicarbonatemia target – bicarbonatemia del paziente)*peso in Kg
In realtà questa formula sebbene indicativa risulta sottostimare il deficit effettivo, in quanto è stato dimostrato che lo spazio di distribuzione del bicarbonato presenta una correlazione inversa rispetto alla bicarbonatemia [95] (full text). Questo fenomeno è dovuto al fatto che, man mano che il sistema bicarbonato va esaurendosi, una maggiore quota di idrogenioni si lega alle proteine. A seguito della somministrazione di bicarbonati, la rigenerazione del sistema tampone lo rende nuovamente disponibile ad accogliere idrogenioni, ed una parte dei bicarbonati somministrati sarà tamponata dai protoni che si trovavano legati alle proteine; questa quota prende il nome di spazio di titolazione. La dose complessiva di bicarbonato dovrebbe essere somministrata in infusione lenta, nel corso di diverse ore piuttosto che in boli, per limitare bruschi aumenti della PaCO2 [79] (full text) [99] [96] [97] [98]. In presenza di una contrazione di volume, se non è presente instabilità emodinamica, la quantità di liquidi somministrati deve essere attentamente valutata al fine di prevenire l’insorgenza di sovraccarico volemico o un’eccessiva alcalinizzazione [28].
Conclusioni
L’acidosi metabolica è di frequente riscontro in clinica, in particolare nel paziente critico e/o con insufficienza renale. È sostenuta da meccanismi complessi la cui conoscenza è indispensabile per effettuare un corretto inquadramento del disordine sottostante. L’acidosi metabolica, soprattutto nella sua forma acuta, può compromettere in maniera significativa l’assetto emodinamico e metabolico dell’organismo, configurando a volte situazioni di effettiva urgenza/emergenza. In tali condizioni è indispensabile un rapido algoritmo diagnostico basato su parametri facilmente ottenibili al letto del malato. L’approccio diagnostico-terapeutico non può prescindere dalla pronta individuazione e correzione di problemi clinici sottostanti, spesso alla base dello squilibrio stesso, come l’ipoperfusione, l’insufficienza respiratoria o l’esposizione a tossici. In assenza di un consenso definitivo sulle indicazioni alla somministrazione della terapia alcalinizzante, la conoscenza dei meccanismi fisiopatologici alla base dello stato di acidosi deve costituire il principio fondante su cui basare i provvedimenti terapeutici che dovranno essere attentamente valutati sulla base dei possibili effetti collaterali, così come l’eventuale ricorso alla terapia sostitutiva della funzione renale. Il trattamento delle acidosi metaboliche ipercloremiche, ad eccezione delle acidosi tubulari renali, costituisce un argomento ancora poco esplorato dalla letteratura, in relazione al quale sono necessari studi che definiscano in maniera più stringente le indicazioni, le modalità e la prognosi associata, soprattutto nel paziente critico.
Bibliografia
[1] Jung B, Rimmele T, Le Goff C et al. Severe metabolic or mixed acidemia on intensive care unit admission: incidence, prognosis and administration of buffer therapy. A prospective, multiple-center study. Critical care (London, England) 2011;15(5):R238 (full text)
[2] Eustace JA, Astor B, Muntner PM et al. Prevalence of acidosis and inflammation and their association with low serum albumin in chronic kidney disease. Kidney international 2004 Mar;65(3):1031-40 (full text)
[3] Gunnerson KJ, Saul M, He S et al. Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients. Critical care (London, England) 2006 Feb;10(1):R22 (full text)
[4] Shane AI, Robert W, Arthur K et al. Acid-base disorders as predictors of early outcomes in major trauma in a resource limited setting: An observational prospective study. The Pan African medical journal 2014 Jan 6;17:2 (full text)
[5] Kellum JA, Song M, Li J et al. Lactic and hydrochloric acids induce different patterns of inflammatory response in LPS-stimulated RAW 264.7 cells. American journal of physiology. Regulatory, integrative and comparative physiology 2004 Apr;286(4):R686-92 (full text)
[7] Besen BA, Gobatto AL, Melro LM et al. Fluid and electrolyte overload in critically ill patients: An overview. World journal of critical care medicine 2015 May 4;4(2):116-29 (full text)
[8] Brunner R, Drolz A, Scherzer TM et al. Renal tubular acidosis is highly prevalent in critically ill patients. Critical care (London, England) 2015 Apr 6;19:148 (full text)
[10] Kraut JA, Kurtz I Toxic alcohol ingestions: clinical features, diagnosis, and management. Clinical journal of the American Society of Nephrology : CJASN 2008 Jan;3(1):208-25 (full text)
[13] Cupisti A, Bottai A, Bellizzi V et al. [Characteristics of patients with chronic kidney disease referred to a nephrology outpatient clinic: results of Nefrodata study]. Giornale italiano di nefrologia : organo ufficiale della Societa italiana di nefrologia 2015 Mar-Apr;32(2) (full text)
[14] Moranne O, Froissart M, Rossert J et al. Timing of onset of CKD-related metabolic complications. Journal of the American Society of Nephrology : JASN 2009 Jan;20(1):164-71 (full text)
[15] Raphael KL, Zhang Y, Wei G et al. Serum bicarbonate and mortality in adults in NHANES III. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association – European Renal Association 2013 May;28(5):1207-13 (full text)
[16] Kovesdy CP, Anderson JE, Kalantar-Zadeh K et al. Association of serum bicarbonate levels with mortality in patients with non-dialysis-dependent CKD. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association – European Renal Association 2009 Apr;24(4):1232-7 (full text)
[17] Raphael KL, Wei G, Baird BC et al. Higher serum bicarbonate levels within the normal range are associated with better survival and renal outcomes in African Americans. Kidney international 2011 Feb;79(3):356-62 (full text)
[21] Wu DY, Shinaberger CS, Regidor DL et al. Association between serum bicarbonate and death in hemodialysis patients: is it better to be acidotic or alkalotic? Clinical journal of the American Society of Nephrology : CJASN 2006 Jan;1(1):70-8 (full text)
[24] Wesson DE, Simoni J Increased tissue acid mediates a progressive decline in the glomerular filtration rate of animals with reduced nephron mass. Kidney international 2009 May;75(9):929-35 (full text)
[25] Frassetto LA, Hsu CY Metabolic acidosis and progression of chronic kidney disease. Journal of the American Society of Nephrology : JASN 2009 Sep;20(9):1869-70 (full text)
[26] Wesson DE, Simoni J Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney international 2010 Dec;78(11):1128-35 (full text)
[27] Kellum JA Clinical review: reunification of acid-base physiology. Critical care (London, England) 2005 Oct 5;9(5):500-7 (full text)
[31] Halperin ML, Kamel KS Some observations on the clinical approach to metabolic acidosis. Journal of the American Society of Nephrology : JASN 2010 Jun;21(6):894-7 (full text)
[34] Boron WF Acid-base transport by the renal proximal tubule. Journal of the American Society of Nephrology : JASN 2006 Sep;17(9):2368-82 (full text)
[37] Kurtz I, Kraut J, Ornekian V et al. Acid-base analysis: a critique of the Stewart and bicarbonate-centered approaches. American journal of physiology. Renal physiology 2008 May;294(5):F1009-31 (full text)
[38] Masevicius FD, Dubin A Has Stewart approach improved our ability to diagnose acid-base disorders in critically ill patients? World journal of critical care medicine 2015 Feb 4;4(1):62-70 (full text)
[43] Chawla LS, Shih S, Davison D et al. Anion gap, anion gap corrected for albumin, base deficit and unmeasured anions in critically ill patients: implications on the assessment of metabolic acidosis and the diagnosis of hyperlactatemia. BMC emergency medicine 2008 Dec 16;8:18 (full text)
[44] Gennari FJ, Weise WJ Acid-base disturbances in gastrointestinal disease. Clinical journal of the American Society of Nephrology : CJASN 2008 Nov;3(6):1861-8 (full text)
[48] Golembiewska E, Ciechanowski K Renal tubular acidosis–underrated problem? Acta biochimica Polonica 2012;59(2):213-7 (full text)
[50] Hall AM, Bass P, Unwin RJ et al. Drug-induced renal Fanconi syndrome. QJM : monthly journal of the Association of Physicians 2014 Apr;107(4):261-9 (full text)
[52] Bouzidi H, Daudon M, Najjar MF et al. [Primary distal renal tubular acidosis]. Annales de biologie clinique 2009 Mar-Apr;67(2):135-40 (full text)
[56] Wesson DE, Simoni J, Broglio K et al. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. American journal of physiology. Renal physiology 2011 Apr;300(4):F830-7 (full text)
[57] Kopple JD, Kalantar-Zadeh K, Mehrotra R et al. Risks of chronic metabolic acidosis in patients with chronic kidney disease. Kidney international. Supplement 2005 Jun;(95):S21-7 (full text)
[61] Kraut JA, Madias NE Lactic acidosis. The New England journal of medicine 2015 Mar 12;372(11):1078-9 (full text)
[63] Suh S Metformin-associated lactic acidosis. Endocrinology and metabolism (Seoul, Korea) 2015 Mar 27;30(1):45-6 (full text)
[64] Kowlgi NG, Chhabra L D-lactic acidosis: an underrecognized complication of short bowel syndrome. Gastroenterology research and practice 2015;2015:476215 (full text)
[66] Soghoian S, Sinert R, Wiener SW et al. Ethylene glycol toxicity presenting with non-anion gap metabolic acidosis. Basic & clinical pharmacology & toxicology 2009 Jan;104(1):22-6 (full text)
[74] Curley GF, Laffey JG Acidosis in the critically ill – balancing risks and benefits to optimize outcome. Critical care (London, England) 2014 Apr 3;18(2):129 (full text)
[75] Velissaris D, Karamouzos V, Ktenopoulos N et al. The Use of Sodium Bicarbonate in the Treatment of Acidosis in Sepsis: A Literature Update on a Long Term Debate. Critical care research and practice 2015;2015:605830 (full text)
[78] Stacpoole PW, Wright EC, Baumgartner TG et al. A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. The New England journal of medicine 1992 Nov 26;327(22):1564-9 (full text)
[79] Sabatini S, Kurtzman NA Bicarbonate therapy in severe metabolic acidosis. Journal of the American Society of Nephrology : JASN 2009 Apr;20(4):692-5 (full text)
[81] Rodríguez-Gutiérrez R, Cámara-Lemarroy CR, Quintanilla-Flores DL et al. Severe Ketoacidosis (pH ≤ 6.9) in Type 2 Diabetes: More Frequent and Less Ominous Than Previously Thought. BioMed research international 2015;2015:134780 (full text)
[82] Glaser N, Barnett P, McCaslin I et al. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. The New England journal of medicine 2001 Jan 25;344(4):264-9 (full text)
[89] Krajewski ML, Raghunathan K, Paluszkiewicz SM et al. Meta-analysis of high- versus low-chloride content in perioperative and critical care fluid resuscitation. The British journal of surgery 2015 Jan;102(1):24-36 (full text)
[92] Dobre M, Rahman M, Hostetter TH et al. Current status of bicarbonate in CKD. Journal of the American Society of Nephrology : JASN 2015 Mar;26(3):515-23 (full text)
[93] Mendoza FJ, Lopez I, Montes de Oca A et al. Metabolic acidosis inhibits soft tissue calcification in uremic rats. Kidney international 2008 Feb;73(4):407-14 (full text)
[94] Lomashvili K, Garg P, O’Neill WC et al. Chemical and hormonal determinants of vascular calcification in vitro. Kidney international 2006 Apr;69(8):1464-70 (full text)
[95] Fernandez PC, Cohen RM, Feldman GM et al. The concept of bicarbonate distribution space: the crucial role of body buffers. Kidney international 1989 Nov;36(5):747-52 (full text)