


Abstract
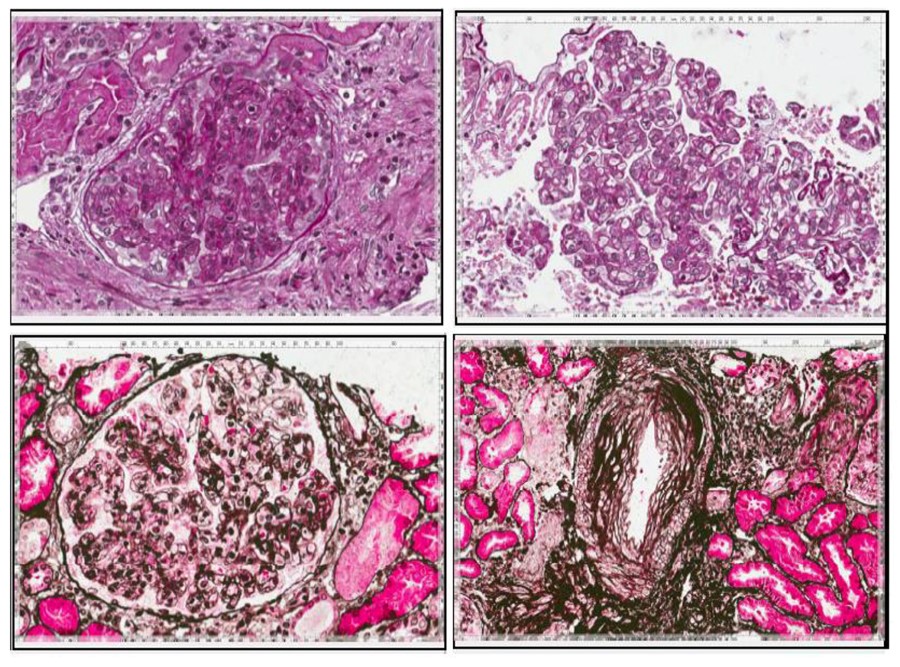
La sindrome di Schnitzler (SS) è un’entità rara e sottodiagnosticata che associa un rash orticarioide cronico, una gammopatia monoclonale IgM (o talvolta IgG) e segni e sintomi di infiammazione sistemica. Negli ultimi 45 anni, la SS è evoluta da un disordine elusivo poco conosciuto al paradigma di una sindrome auto-infiammatoria acquisita tardiva. Sebbene non vi sia una prova certa della sua precisa patogenesi, dovrebbe essere considerata una malattia acquisita che comporta una stimolazione anormale del sistema immunitario innato, che può essere annullata dall’antagonista del recettore dell’interleuchina 1 (IL-1) anakinra. Qui descriviamo il caso di un paziente caucasico maschio di 56 anni affetto da SS e ospedalizzato più volte nella nostra unità a causa di episodi recidivanti di insufficienza renale acuta. È stato sottoposto a biopsia renale percutanea eco-guidata nel settembre 2012, che ha mostrato il quadro istologico della glomerulonefrite membranoproliferativa di tipo I. È stato sottoposto a terapie convenzionali, tra cui farmaci anti-infiammatori non steroidei, steroidi e farmaci immunosoppressivi; più recentemente è stato prescritto l’antagonista del recettore dell’IL-1 anakinra, con notevole beneficio clinico. Sebbene la letteratura riguardante il coinvolgimento dei reni nella SS sia davvero scarsa, esso può essere tuttavia così grave, come nel caso qui riportato, da farci raccomandare la ricerca sistematica dei marcatori di nefropatia nella SS.
Parole Chiave: Insufficienza renale acuta, malattie autoinfiammatorie, eruzione urticarioide cronica, glomerulonefrite membranoproliferativa, gammopatia monoclonale IgM, sindrome di Schnitzler
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}