


Abstract
Introduzione: In Italia e nel resto del mondo sono state effettuate numerose indagini epidemiologiche relative alla prevalenza/incidenza della sindrome di Fabry (SF) e su altre malattie ad accumulo lisosomiale (LSDs). Le survey epidemiologiche sottostimano ancora la reale prevalenza della SF e delle sue numerose varianti genetiche. La distribuzione varia a seconda delle aree geografiche, e/o delle varie etnie. Oggi anche le donne eterozigoti possono essere colpite da forme severe e pluri-sintomatiche della SF.
Scopo: Abbiamo iniziato un’indagine su tutta la popolazione di nefropatici seguiti in circa 20 ambulatori territoriali e nei pazienti in dialisi senza diagnosi certa all’interno della nostra area sanitaria composta da 319.340 abitanti.
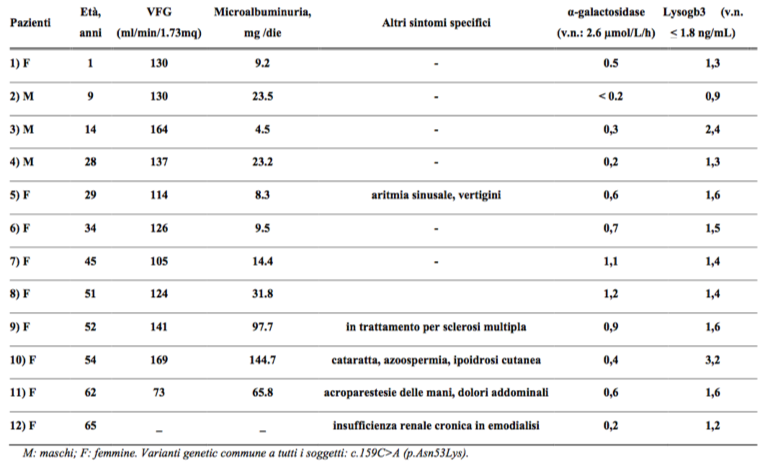
Metodi: I pazienti selezionati dai nostri ambulatori sono sino ad oggi 2.710, di questi 150 pazienti sono risultati sospetti (73 in dialisi e 77 in trattamento conservativo). Una paziente di sesso femminile in dialisi è risultata positiva; pertanto abbiamo ritenuto indagare su tutti i suoi collaterali rintracciabili individuando così altri 11 pazienti (totale: 4 maschi e 7 femmine). Tutti questi pazienti risiedono in una microarea di 21.822 abitanti. E’ risultata così una prevalenza di un caso positivo su 1.818 abitanti. Questi dati si riferiscono ai 18 mesi iniziali di screening.
Conclusioni: Nei pazienti con una proteinuria o una microalbuminuria paranormale (150-200 mg/die) appare doveroso effettuare lo screening per la SF specialmente nelle aree con una non chiara alta incidenza e/o prevalenza di nefropatie. Una volta individuati i pazienti positivi di ambo i sessi, in questi dovrebbero essere poi rapidamente valutati gli organi ed apparati più colpiti dalla SF e quindi condurre un’accurata valutazione cardiologica e neurologica.
PAROLE CHIAVE: malattia di Fabry, malattie rare, patrimonio genetico
{kind=link}
{kind=link}
{kind=link}
{kind=link}