


Abstract
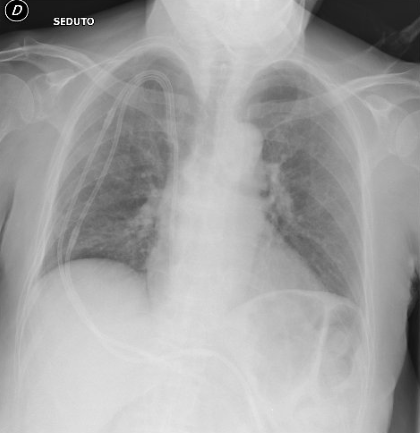
La poliangioite granulomatosa è una vasculite ANCA-relata (AAV) le cui manifestazioni cliniche riguardano principalmente il tratto respiratorio (superiore ed inferiore) ed il rene. Il trattamento della poliangioite granulomatosa (così come delle AAV in generale) concerne l’utilizzo di farmaci immunosoppressori, non scevri da effetti collaterali; le complicanze più frequenti sono quelle infettive e quelle neoplastiche. La malattia presenta spesso recidive. L’EBV è un virus ubiquitario; si stima che circa il 90% della popolazione mondiale sia entrato in contatto con tale patogeno ed abbia sviluppato successivamente un’infezione latente. In alcune condizioni, tra cui l’immunosoppressione, il virus dell’EBV può riattivarsi. Riportiamo il caso clinico di una donna di 67 anni affetta da GPA esordita con coinvolgimento del tratto respiratorio superiore e renale, con esito in insufficienza renale con necessità di trattamento emodialitico; in corso di terapia di induzione di remissione (prednisone e ciclofosfamide), si presentava presso il nostro reparto con un quadro di dispnea acuta ed insufficienza respiratoria. Esclusa l’embolia polmonare e lo scompenso cardiaco, sono state effettuate una serie di indagini radiologiche (HRTC) e strumentali (fibronscopia con BAL) che hanno escluso la presenza di reperti suggestivi di riattivazione polmonare di vasculite (in prima linea veniva esclusa un’emorragia alveolare diffusa). Si evidenziava la presenza al BAL di EBV-DNA. Veniva indi posta diagnosi di polmonite da EBV e si intraprendeva terapia antivirale con Aciclovir, con miglioramento clinico e radiologico del quadro. Nella GPA, in paziente in terapia immunosoppressiva, un coinvolgimento polmonare può essere dovuto alla malattia di base, tuttavia va sempre esclusa una causa infettiva, anche da agenti atipici.
Parole chiave: polmonite da EBV, poliangioite granulomatosa, aciclovir
{kind=link}
{kind=link}